Your new post is loading...

|
Scooped by
Juan Lama
|
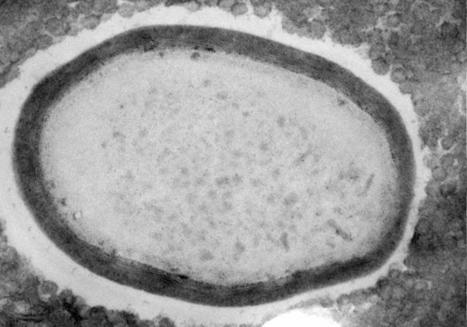
Genetic analysis of 50,000-year-old Neanderthal skeletons has uncovered the remnants of three viruses related to modern human pathogens, and the researchers think they could be recreated. Genetic sequences from three common viruses that plague humanity today have been isolated from the remains of Neanderthals who lived more than 50,000 years ago. Marcelo Briones at the Federal University of São Paulo, Brazil, says it may be possible to synthesise these viruses and infect modern human cells with them in the lab... Preprint of the study in bioRxiv: https://doi.org/10.1101/2023.03.16.532919
|
|
Scooped by
Juan Lama
|
Introduction Continued SARS-CoV-2 infection among immunocompromised individuals is likely to play a role in generating genomic diversity and the emergence of novel variants. Antiviral treatments such as molnupiravir are used to mitigate severe COVID-19 outcomes, but the extended effects of these drugs on viral evolution in patients with chronic infections remain uncertain. This study investigates how molnupiravir affects SARS-CoV-2 evolution in immunocompromised patients with prolonged infections. Methods The study included five immunocompromised patients treated with molnupiravir and four patients not treated with molnupiravir (two immunocompromised and two non-immunocompromised). We selected patients who had been infected by similar SARS-CoV-2 variants and with high-quality genomes across timepoints to allow comparison between groups. Throat and nasopharyngeal samples were collected in patients up to 44 days post treatment and were sequenced using tiled amplicon sequencing followed by variant calling. The UShER pipeline and University of California Santa Cruz genome viewer provided insights into the global context of variants. Treated and untreated patients were compared, and mutation profiles were visualised to understand the impact of molnupiravir on viral evolution. Findings Patients treated with molnupiravir showed a large increase in low-to-mid-frequency variants in as little as 10 days after treatment, whereas no such change was observed in untreated patients. Some of these variants became fixed in the viral population, including non-synonymous mutations in the spike protein. The variants were distributed across the genome and included unique mutations not commonly found in global omicron genomes. Notably, G-to-A and C-to-T mutations dominated the mutational profile of treated patients, persisting up to 44 days post treatment. Interpretation Molnupiravir treatment in immunocompromised patients led to the accumulation of a distinctive pattern of mutations beyond the recommended 5 days of treatment. Treated patients maintained persistent PCR positivity for the duration of monitoring, indicating clear potential for transmission and subsequent emergence of novel variants. Published in The Lancet Microbe (March 22, 2024):
|
|
Scooped by
Juan Lama
|
Study finds deer are virus reservoirs, promoting ongoing mutation. New research has found that white-tailed deer across Ohio have been infected with the virus that causes COVID-19. Alarmingly, the results also show that viral variants evolve about three times faster in deer than in humans. Scientists collected 1,522 nasal swabs from free-ranging deer in 83 of the state’s 88 counties between November 2021 and March 2022. More than 10% of the samples were positive for the SARS-CoV-2 virus, and at least one positive case was found in 59% of the counties in which testing took place. Genomic analysis showed that at least 30 infections in deer had been introduced by humans – a figure that surprised the research team. “We generally talk about interspecies transmission as a rare event, but this wasn’t a huge sampling, and we’re able to document 30 spillovers. It seems to be moving between people and animals quite easily,” said Andrew Bowman, associate professor of veterinary preventive medicine at The Ohio State University and co-senior author of the study. “And the evidence is growing that humans can get it from deer – which isn’t radically surprising. It’s probably not a one-way pipeline.” The combined findings suggest that the white-tailed deer species is a reservoir for SARS-CoV-2 that enables continuing mutation, and that the virus’s circulation in deer could lead to its spread to other wildlife and livestock. The study is published today (August 28, 2023) in the journal Nature Communications. Previous Observations and Expansions Bowman and colleagues previously reported detection of SARS-CoV-2 infections in white-tailed deer in nine Ohio locations in December 2021, and are continuing to monitor deer for infection by more recent variants. “We expanded across Ohio to see if this was a localized problem – and we find it in lots of places, so it’s not just a localized event,” Bowman said. “Some of the thought back then was that maybe it’s just in urban deer because they’re in closer contact with people. But in rural parts of the state, we’re finding plenty of positive deer.” Beyond the detection of active infections, researchers also found through blood samples containing antibodies – indicating previous exposure to the virus – that an estimated 23.5% of deer in Ohio had been infected at one time or another. Variant Analysis The 80 whole-genome sequences obtained from the collected samples represented groups of various viral variants: the highly contagious delta variant, the predominant human strain in the United States in the early fall of 2021 that accounted for almost 90% of the sequences, and alpha, the first named variant of concern that had circulated in humans in the spring of 2021. The analysis revealed that the genetic composition of delta variants in deer matched dominant lineages found in humans at the time, pointing to the spillover events, and that deer-to-deer transmission followed in clusters, some spanning multiple counties. “There’s probably a timing component to what we found – we were near the end of a delta peak in humans, and then we see a lot of delta in deer,” Bowman said. “But we were well past the last alpha detection in humans. So the idea that deer are holding onto lineages that have since gone extinct in humans is something we were worried about.” The study did suggest that COVID-19 vaccination is likely to help protect people against severe disease in the event of a spillover back to humans. An analysis of the effects of deer variants on Siberian hamsters, an animal model for SARS-CoV-2 studies, showed that vaccinated hamsters did not get as sick from infection as unvaccinated animals. Rapid Evolution in Deer Disturbingly, the variants circulating in deer are expected to continue to change. An investigation of the mutations found in the samples provided evidence of more rapid evolution of both alpha and delta variants in deer compared to humans. “Not only are deer getting infected with and maintaining SARS-CoV-2, but the rate of change is accelerated in deer – potentially away from what has infected humans,” Bowman said. How the virus is transmitted from humans to white-tailed deer remains a mystery. And so far, even with about 30 million free-ranging deer in the U.S., no substantial outbreaks of deer-origin strains have occurred in humans. Potential Implications Circulation among animals, however, remains highly likely. Bowman noted that about 70% of free-ranging deer in Ohio have not been infected or exposed to the virus, “so that’s a large body of naive animals that the virus could spread through rather uninhibited.” “Having that animal host in play creates things we need to watch out for,” he said. “If this trajectory continues for years and we have a virus that becomes deer-adapted, then does that become the pathway into other animal hosts, wildlife or domestic? We just don’t know.” Martha Nelson of the National Library of Medicine was co-corresponding author of the study. Ohio State co-authors Dillon McBride, Steven Overend, Devra Huey, Amanda Williams, Seth Faith and Jacqueline Nolting worked on the study with co-authors from St. Jude Children’s Research Hospital; the University of California, Los Angeles; the National Research Centre in Giza, Egypt; PathAI Diagnostics; the Ohio Department of Natural Resources; the U.S. Department of Agriculture; Columbus and Franklin County Metroparks; and the Rega Institute for Medical Research in Belgium. Cited Study published in Nat. Communications (Aug. 28, 2023) https://doi.org/10.1038/s41467-023-40706-y
|
|
Scooped by
Juan Lama
|
Corals, sturgeon and other aquatic creatures harbour signs of infection by influenza and its distant relatives. The influenza virus might have started in fish. Researchers trawling genetic databases have discovered a distant relative of influenza viruses — which are responsible for seasonal flu, not to mention the avian flu roiling the globe — in sturgeon1. The authors also found that the wider virus family that includes influenza probably originated hundreds of millions of years ago in primordial aquatic animals that evolved well before the first fish. Viruses in this group seem to be especially adept at jumping between hosts, says Mary Petrone, a virologist at the University of Sydney, Australia, who co-authored the preprint describing the findings. Knowing about ancient host jumps could help scientists identify viruses with the potential to spark new human epidemics. The study was posted on 16 Feburary to the preprint server bioRxiv and has not yet been peer reviewed. Influenza’s origins story Like many virologists, Petrone spent the first couple years of the pandemic intensively studying SARS-CoV-2. But when she moved to Australia to do postdoctoral research, Petrone wanted to steer clear of human infections and spend time in one of the country’s most famous ecosystems. “After working on COVID for two years, I thought going to coral reefs to do fieldwork sounded really good,” she says. Corals are part of a phylum called Cnidaria, whose ancestors branched off from other animals around 600 million years ago. Petrone hoped that studying corals could reveal the deeper history of viruses that infect animals — particularly those with RNA genomes. This viral group includes numerous human and animal pathogens. Petrone’s first call was not to a diving shop but to Zoe Richards, a coral-reef researcher at Curtin University in Bentley, Australia, who provided samples of two coral species collected off the coast of Western Australia. Analysis of RNA collected from the corals found evidence of infection with viruses that belong to a grouping called Articulavirales, which includes influenza’s family of viruses and a group called Quaranjaviruses. The latter group’s members circulate in ticks and occasionally spill over into humans, birds and other vertebrates. The new analysis suggests that coral-infecting viruses are part of an ancient viral family that probably emerged around 600 million years ago, and later gave rise to other members of Articulavirales, including influenza and Quaranjaviruses. Secrets of the hagfish The discovery got Petrone wondering whether influenza viruses might also have been born at sea. There was already some evidence for this. In 2018, researchers identified a distant relative of influenza in hagfish2. These slimy, jawless creatures descended from an early lineage of vertebrates, and the study’s authors hinted that influenza evolved alongside vertebrates. Searching genetic databases, Petrone found influenza-related RNA sequences in samples from Siberian sturgeon (Acipenser baerii). Sturgeon are jawed vertebrates, more closely related to humans than hagfish are. But the sturgeon virus had branched off from the main influenza family tree before any other known influenza virus, including the hagfish virus. The discovery of the two early lineages of influenza suggest that influenza probably infected aquatic animals, including fish, before moving onto land, says Petrone. But it’s not clear whether influenza moved onto land with early terrestrial vertebrates, or jumped from sea to land more recently. To determine this, researchers will need to look for relatives of influenza in more animals and gain a better understand how the virus spreads between host species, researchers say. Born at sea Jie Cui, an evolutionary virologist at the Pasteur Institute of Shanghai in China, agrees that influenza and its wider family probably emerged from the sea. In 2021, his team analysed deep-sea lobster genomes and identified viruses that are part of influenza’s wider group3. “There is great untapped viral diversity in aquatic environments,” he says. Robert Gifford, an evolutionary virologist at the University of Glasgow, UK, says it would be surprising to find a major group of viruses that didn’t arise in aquatic environments because of the ancient nature of marine life. “The study provides compelling evidence that influenza viruses have an aquatic origin.” Identifying ancient host jumps could also help researchers gauge the risk that certain viruses pose to humans, researchers say. Petrone’s team found signs that Quaranjaviruses that infect ticks might have jumped to the creatures after first circulating in crustaceans. Uncovering such jumps shows that the study of aquatic viruses “can help us to better understand the historic emergence and evolution of viruses with zoonotic potential”, adds Chantal Vogels, an arbovirologist at the Yale School of Public Health in New Haven, Connecticut. Gifford agrees that studies such as Petrone’s could help to identify viruses that have the capacity to spark epidemics in humans and other animals. But he cautions that conclusions about ancient host jumps can change as more of the viral family tree gets filled in, reshuffling relationships. Cited research available in bioRxiv (Feb. 16, 2023): https://doi.org/10.1101/2023.02.15.528772
|
|
Scooped by
Juan Lama
|
The geographic and evolutionary origins of the SARS-CoV-2 Omicron variant (BA.1), which was first detected mid-November 2021 in Southern Africa, remain unknown. We tested 13,097 COVID-19 patients sampled between mid-2021 to early 2022 from 22 African countries for BA.1 by real-time RT-PCR. By November-December 2021, BA.1 had replaced the Delta variant in all African sub-regions following a South-North gradient, with a peak Rt of 4.1. Polymerase chain reaction and near-full genome sequencing data revealed genetically diverse Omicron ancestors already existed across Africa by August 2021. Mutations, altering viral tropism, replication and immune escape, gradually accumulated in the spike gene. Omicron ancestors were therefore present in several African countries months before Omicron dominated transmission. These data also indicate that travel bans are ineffective in the face of undetected and widespread infection. Published in Science (Dec. 1, 2022): https://doi.org/10.1126/science.add8737
|
|
Scooped by
Juan Lama
|
Significance Tracking the animal reservoir of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its variants is important for understanding the current COVID-19 pandemic and preventing future pandemics. Speculations about the source of the omicron variant are abundant, yet experimental evidence has been scarce. Here, we provide the structural information on how omicron recognizes its mouse receptor. Our study demonstrates that the omicron mutations in the receptor-binding region are structurally adapted to mouse angiotensin-converting enzyme 2 (ACE2), informing an understanding of the origin of the omicron variant and the evolution of SARS-CoV-2. It may facilitate epidemiological surveillance of SARS-CoV-2 in animals to prevent future coronavirus pandemics. Abstract The sudden emergence and rapid spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) omicron variant has raised questions about its animal reservoir. Here, we investigated receptor recognition of the omicron’s receptor-binding domain (RBD), focusing on four of its mutations (Q493R, Q498R, N501Y, and Y505H) surrounding two mutational hotspots. These mutations have variable effects on the RBD’s affinity for human angiotensin-converting enzyme 2 (ACE2), but they all enhance the RBD’s affinity for mouse ACE2. We further determined the crystal structure of omicron RBD complexed with mouse ACE2. The structure showed that all four mutations are viral adaptations to mouse ACE2: three of them (Q493R, Q498R, and Y505H) are uniquely adapted to mouse ACE2, whereas the other one (N501Y) is adapted to both human ACE2 and mouse ACE2. These data reveal that the omicron RBD was well adapted to mouse ACE2 before omicron started to infect humans, providing insight into the potential evolutionary origin of the omicron variant. Published in PNAS (Oct.18, 2022):
|
|
Scooped by
Juan Lama
|
Long-term SARS-CoV-2 infections in immunodeficient patients are an important source of variation for the virus but are understudied. Many case studies have been published which describe one or a small number of long-term infected individuals but no study has combined these sequences into a cohesive dataset. This work aims to rectify this and study the genomics of this patient group through a combination of literature searches as well as identifying new case series directly from the COG-UK dataset. The spike gene receptor binding domain (RBD) and N-terminal domains (NTD) were identified as mutation hotspots. Numerous mutations associated with variants of concern were observed to emerge recurrently. Additionally a mutation in the envelope gene, - T30I was determined to be the most recurrent frequently occurring mutation arising in persistent infections. A high proportion of recurrent mutations in immunodeficient individuals are associated with ACE2 affinity, immune escape, or viral packaging optimisation. There is an apparent selective pressure for mutations which aid intra-host transmission or persistence which are often different to mutations which aid inter-host transmission, although the fact that multiple recurrent de novo mutations are considered defining for variants of concern strongly indicates that this potential source of novel variants should not be discounted. Preprint available in medRxiv (March 2, 2022): https://doi.org/10.1101/2022.03.02.22271697
|
|
Scooped by
Juan Lama
|
A few dozen human genes rapidly evolved in ancient East Asia to thwart coronavirus infections, scientists say. Those genes could be crucial to today’s pandemic. Researchers have found evidence that a coronavirus epidemic swept East Asia some 20,000 years ago and was devastating enough to leave an evolutionary imprint on the DNA of people alive today. The new study suggests that an ancient coronavirus plagued the region for many years, researchers say. The finding could have dire implications for the Covid-19 pandemic if it’s not brought under control soon through vaccination. “It should make us worry,” said David Enard, an evolutionary biologist at the University of Arizona who led the study, which was published on Thursday in the journal Current Biology. “What is going on right now might be going on for generations and generations.” Until now, researchers could not look back very far into the history of this family of pathogens. Over the past 20 years, three coronaviruses have adapted to infect humans and cause severe respiratory disease: Covid-19, SARS and MERS. Studies on each of these coronaviruses indicate that they jumped into our species from bats or other mammals. Four other coronaviruses can also infect people, but they usually cause only mild colds. Scientists did not directly observe these coronaviruses becoming human pathogens, so they have relied on indirect clues to estimate when the jumps happened. Coronaviruses gain new mutations at a roughly regular rate, and so comparing their genetic variation makes it possible to determine when they diverged from a common ancestor. The most recent of these mild coronaviruses, called HCoV-HKU1, crossed the species barrier in the 1950s. The oldest, called HCoV-NL63, may date back as far as 820 years. But before that point, the coronavirus trail went cold — until Dr. Enard and his colleagues applied a new method to the search. Instead of looking at the genes of the coronaviruses, the researchers looked at the effects on the DNA of their human hosts. Over generations, viruses drive enormous amounts of change in the human genome. A mutation that protects against a viral infection may well mean the difference between life and death, and it will be passed down to offspring. A lifesaving mutation, for example, might allow people to chop apart a virus’s proteins. But viruses can evolve, too. Their proteins can change shape to overcome a host’s defenses. And those changes might spur the host to evolve even more counteroffensives, leading to more mutations. When a random new mutation happens to provide resistance to a virus, it can swiftly become more common from one generation to the next. And other versions of that gene, in turn, become rarer. So if one version of a gene dominates all others in large groups of people, scientists know that is most likely a signature of rapid evolution in the past. In recent years, Dr. Enard and his colleagues have searched the human genome for these patterns of genetic variation in order to reconstruct the history of an array of viruses. When the pandemic struck, he wondered whether ancient coronaviruses had left a distinctive mark of their own. He and his colleagues compared the DNA of thousands of people across 26 different populations around the world, looking at a combination of genes known to be crucial for coronaviruses but not other kinds of pathogens. In East Asian populations, the scientists found that 42 of these genes had a dominant version. That was a strong signal that people in East Asia had adapted to an ancient coronavirus. But whatever happened in East Asia seemed to have been limited to that region. “When we compared them to populations around the world, we couldn’t find the signal,” said Yassine Souilmi, a postdoctoral researcher at the University of Adelaide in Australia and a co-author of the new study. The scientists then tried to estimate how long ago East Asians had adapted to a coronavirus. They took advantage of the fact that once a dominant version of a gene starts being passed down through the generations, it can gain harmless random mutations. As more time passes, more of those mutations accumulate. Dr. Enard and his colleagues found that the 42 genes all had about the same number of mutations. That meant that they had all rapidly evolved at about the same time. “This is a signal we should absolutely not expect by chance,” Dr. Enard said. They estimated that all of those genes evolved their antiviral mutations sometime between 20,000 and 25,000 years ago, most likely over the course of a few centuries. It’s a surprising finding, since East Asians at the time were not living in dense communities but instead formed small bands of hunter-gatherers. Aida Andres, an evolutionary geneticist at the University College London who was not involved in the new study, said she found the work compelling. “I’m quite convinced there’s something there,” she said. Still, she didn’t think it was possible yet to make a firm estimate of how long ago the ancient epidemic took place. “The timing is a complicated thing,” she said. “Whether that happened a few thousand years before or after — I personally think it’s something that we cannot be as confident of.” Scientists looking for drugs to fight the new coronavirus might want to scrutinize the 42 genes that evolved in response to the ancient epidemic, Dr. Souilmi said. “It’s actually pointing us to molecular knobs to adjust the immune response to the virus,” he said. Dr. Anders agreed, saying that the genes identified in the new study should get special attention as targets for drugs. “You know that they’re important,” she said. “That’s the nice thing about evolution.”
|
|
Scooped by
Juan Lama
|
Brazil's P1 coronavirus variant, behind a deadly COVID-19 surge in the Latin American country that has raised international alarm, is mutating in ways that could make it better able to evade antibodies, according to scientists studying the virus. Research conducted by the public health institute Fiocruz into the variants circulating in Brazil found mutations in the spike region of the virus that is used to enter and infect cells. Those changes, the scientists said, could make the virus more resistant to vaccines - which target the spike protein - with potentially grave implications for the severity of the outbreak in Latin America’s most populous nation. “We believe it’s another escape mechanism the virus is creating to evade the response of antibodies,” said Felipe Naveca, one of the authors of the study and part of Fiocruz in the Amazon city of Manaus, where the P1 variant is believed to have originated. Naveca said the changes appeared to be similar to the mutations seen in the even more aggressive South African variant, against which studies have shown some vaccines have substantially reduced efficacy. “This is particularly worrying because the virus is continuing to accelerate in its evolution,” he added. Studies have shown the P1 variant to be as much as 2.5 times more contagious than the original coronavirus and more resistant to antibodies. On Tuesday, France suspended all flights to and from Brazil in a bid to prevent the variant’s spread as Latin America’s largest economy becomes increasingly isolated. The variant, which has quickly become dominant in Brazil, is thought to be a large factor behind a massive second wave that has brought the country’s death toll to over 350,000 - the second highest in the world behind the United States. Brazil’s outbreak is also increasingly affecting younger people, with hospital data showing that in March more than half of all patients in intensive care were aged 40 or younger. For Ester Sabino, a scientist at the faculty of medicine of the University of Sao Paulo who led the first genome sequencing of the coronavirus in Brazil, the mutations of the P1 variant are not surprising given the fast pace of transmission. “If you have a high level of transmission, like you have in Brazil at the moment, your risk of new mutations and variants increases,” she said. Fiocruz researchers including Naveca also recently described a novel variant descended from a different lineage to P1, and detected in the northeast of the country, that carried 14 defining mutations including the E484K change first noted in the South African variant (bit.ly/2RBhLKD).
|
|
Scooped by
Juan Lama
|
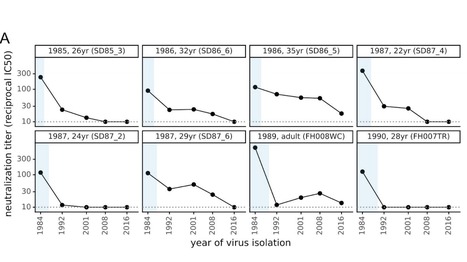
There is intense interest in antibody immunity to coronaviruses. However, it is unknown if coronaviruses evolve to escape such immunity, and if so, how rapidly. Here we address this question by characterizing the historical evolution of human coronavirus 229E. We identify human sera from the 1980s and 1990s that have neutralizing titers against contemporaneous 229E that are comparable to the anti-SARS-CoV-2 titers induced by SARS-CoV-2 infection or vaccination. We test these sera against 229E strains isolated after sera collection, and find that neutralizing titers are lower against these "future" viruses. In some cases, sera that neutralize contemporaneous 229E viral strains with titers >1:100 do not detectably neutralize strains isolated 8-17 years later. The decreased neutralization of "future" viruses is due to antigenic evolution of the viral spike, especially in the receptor-binding domain. If these results extrapolate to other coronaviruses, then it may be advisable to periodically update SARS-CoV-2 vaccines. Preprint available in bioRxiV (Dec. 18, 2020): https://doi.org/10.1101/2020.12.17.423313
|
|
Scooped by
Juan Lama
|
Similar to bacteria evolving resistance to antibiotics, viruses can evolve resistance to vaccines, and the evolution of SARS-CoV-2 could undermine the effectiveness of vaccines that are currently under development, according to a paper published November 9 in the open-access journal PLOS Biology by David Kennedy and Andrew Read from Pennsylvania State University, U.S. The authors also offer recommendations to vaccine developers for minimizing the likelihood of this outcome. "A COVID-19 vaccine is urgently needed to save lives and help society return to its pre-pandemic normal," said David Kennedy, assistant professor of biology. "As we have seen with other diseases, such as pneumonia, the evolution of resistance can quickly render vaccines ineffective. By learning from these previous challenges and by implementing this knowledge during vaccine design, we may be able to maximize the long-term impact of COVID-19 vaccines." The researchers specifically suggest that the standard blood and nasal-swab samples taken during clinical trials to quantify individuals' responses to vaccination may also be used to assess the likelihood that the vaccines being tested will drive resistance evolution. For example, the team proposes that blood samples can be used to assess the redundancy of immune protection generated by candidate vaccines by measuring the types and amounts of antibodies and T-cells that are present. "Much like how combination antibiotic therapy delays the evolution of antibiotic resistance, vaccines that are designed to induce a redundant immune response—or one in which the immune system is encouraged to target multiple sites, called epitopes—on the virus's surface, can delay the evolution of vaccine resistance," said Andrew Read, Evan Pugh Professor of Biology and Entomology and director of the Huck Institutes of the Life Sciences. "That's because the virus would have to acquire several mutations, as opposed to just one, in order to survive the host immune system's attack." The researchers also recommend that nasal swabs typically collected during clinical trials may be used to determine the viral titer, or amount of virus present, which can be considered a proxy for transmission potential. They noted that strongly suppressing virus transmission through vaccinated hosts is key to slowing the evolution of resistance, since it minimizes opportunities for mutations to arise and reduces opportunities for natural selection to act on those mutations that do arise. In addition, the team suggests that the genetic data acquired through nasal swabs can be used to examine whether vaccine-driven selection has occurred. For example, differences in alleles, or forms of genes that arise from mutations, between the viral genomes collected from vaccinated versus unvaccinated individuals would indicate that selection has taken place. "According to the World Health Organization, at least 198 COVID-19 vaccines are in the development pipeline, with 44 currently undergoing clinical evaluation," said Kennedy. "We suggest that the risk of resistance be used to prioritize investment among otherwise similarly promising vaccine candidates." Publication cited available in PLOS Biology (Nov. 9 , 2020): https://doi.org/10.1371/journal.pbio.3001000
|
|
Scooped by
Juan Lama
|
This is a phylogenetic network of SARS-CoV-2 genomes sampled from across the world. These genomes are closely related and under evolutionary selection in their human hosts, sometimes with parallel evolution events, that is, the same virus mutation emerges in two different human hosts. This makes character-based phylogenetic networks the method of choice for reconstructing their evolutionary paths and their ancestral genome in the human host. The network method has been used in around 10,000 phylogenetic studies of diverse organisms, and is mostly known for reconstructing the prehistoric population movements of humans and for ecological studies, but is less commonly employed in the field of virology.... The described core mutations have been confirmed by a variety of contributing laboratories and sequencing platforms and can be considered reliable. The phylogeographic patterns in the network are potentially affected by distinctive migratory histories, founder events, and sample size. Nevertheless, it would be prudent to consider the possibility that mutational variants might modulate the clinical presentation and spread of the disease. The phylogenetic classification provided here may be used to rule out or confirm such effects when evaluating clinical and epidemiological outcomes of SARS-CoV-2 infection, and when designing treatment and, eventually, vaccines.
|
|
Scooped by
Juan Lama
|
Many animals, including humans, have DNA left over from ancient viral infections. In koalas, researchers are studying the process in real time. Koalas have been running into hard times. They have suffered for years from habitat destruction, dog attacks, automobile accidents. But that’s only the beginning. They are also plagued by chlamydia and cancers like leukemia and lymphoma, and in researching those problems, scientists have found a natural laboratory in which to study one of the hottest topics in biology: how viruses can insert themselves into an animal’s DNA and sometimes change the course of evolution. The target of this research is Koala retrovirus, or KoRV, a bit of protein and genetic material in the same family as H.I.V. that began inserting itself into the koala genome about 40,000 years ago and is now passed on from generation to generation, like genes. It is also still passed from animal, as a typical viral infection. In recent years, scientists have found that the insertion of viruses into the genomes of animals has occurred over and over again. An estimated 8 percent of the human genome is made up of viruses left over from ancient infections, ancient as in millions of years ago, many of them in primate ancestors before human beings existed. The koala retrovirus is unusual because 40,000 years is the blink of an eye in evolutionary time, and because the process appears to be continuing. A group of scientists reported in Cell on Thursday that they observed a genome immune system fighting to render the virus inactive now that it has established itself in the koala DNA. They also reported that koala retrovirus may have activated other ancient viral DNA. All of this activity stirs the pot of mutation and variation that is the raw material for natural selection. Koala genetics are a gold mine, said William Theurkauf, a professor in molecular medicine at the University of Massachusetts Medical School and one of the authors of the report. “What they are going through is the process of what’s driven the evolution of every animal on the planet.” Past viral infections have led to major evolutionary changes, he said. For example: “A gene that is absolutely essential for the placenta was derived from the shell of a virus millions of years ago.” Humans would not exist without that ancient retroviral infection. Retroviruses are made of RNA, a single strand of genetic information. When they infect a cell, they translate themselves into DNA, the two-stranded molecule that carries all the information for making humans, koalas and other animals. The retroviruses take over the DNA machinery to make more of themselves, which keeps the process going. That process makes us and other animals sick. AIDS is probably the best known retroviral disease. But when the insertion of a retrovirus occurs in a sperm or an egg cell, the change can become permanent, passed on forever. When retroviruses become part of an animal’s inherited DNA, they are called endogenous and eventually they no longer cause the kind of original infection they once did. But they can still be used by the animal's genetic machinery for other purposes, like making a placenta... Additional information published in Cell on October 10, 2019: https://doi.org/10.1016/j.cell.2019.09.002
|
|
|
Scooped by
Juan Lama
|
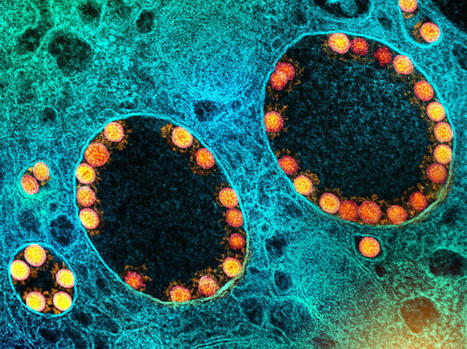
Large and giant double-stranded DNA viruses within the phylum Nucleocytoviricota are diverse and prevalent in the environment where they substantially affect the ecology and evolution of eukaryotes. Until now, these viruses were only sporadically found in the digestive system of vertebrates. Here, we present the identification and genomic characterization of a proposed third order of viruses within the class Pokkesviricetes that currently consists of poxviruses and asfuviruses. Members of this newly identified order we provisionally named Egovirales are abundant in the digestive system of vertebrates worldwide and occur in high abundances in >10% of livestock animals, >2% of humans, and wild animals. Egoviruses have linear genomes up to 360 kbp in length that likely produce multilayered icosahedral capsids, similar to those of asfuviruses. The diversity of egoviruses already far exceeds that of all known poxviruses and animal-associated asfuviruses. Phylogenetic analyses and patterns of virus distribution across vertebrates suggest that egoviruses can be either specialists or generalists associated with a single or multiple vertebrate species, respectively. Notably, one egovirus clade is human-specific, evolutionarily constrained, and spread across continents, demonstrating a long-lasting association between Egovirales and the human population on the global scale. Egoviruses not only expand the ecological and evolutionary scope of Pokkesviricetes, but also appear to be the most diverse, widespread, and abundant group of double-stranded DNA viruses infecting eukaryotic cells in the digestive system of vertebrates. Preprint in bioRxiv (March 23, 2024): https://doi.org/10.1101/2024.03.23.586382
|
|
Scooped by
Juan Lama
|
Background The COVID-19 pandemic, caused by the Severe Acute Respiratory Syndrome Coronavirus 2 virus, emerged in late 2019 and spready globally. Many effects of infection with this pathogen are still unknown, with both chronic and repeated COVID-19 infection producing novel pathologies. Case presentation An immunocompromised patient presented with chronic COVID-19 infection. The patient had history of Hodgkin’s lymphoma, treated with chemotherapy and stem cell transplant. During the course of their treatment, eleven respiratory samples from the patient were analyzed by whole-genome sequencing followed by lineage identification. Whole-genome sequencing of the virus present in the patient over time revealed that the patient at various timepoints harboured three different lineages of the virus. The patient was initially infected with the B.1.1.176 lineage before coinfection with BA.1. When the patient was coinfected with both B.1.1.176 and BA.1, the viral populations were found in approximately equal proportions within the patient based on sequencing read abundance. Upon further sampling, the lineage present within the patient during the final two timepoints was found to be BA.2.9. The patient eventually developed respiratory failure and died. Conclusions This case study shows an example of the changes that can happen within an immunocompromised patient who is infected with COVID-19 multiple times. Furthermore, this case demonstrates how simultaneous coinfection with two lineages of COVID-19 can lead to unclear lineage assignment by standard methods, which are resolved by further investigation. When analyzing chronic COVID-19 infection and reinfection cases, care must be taken to properly identify the lineages of the virus present. Key points -
A patient repeatedly tested positive for COVID-19 over 16 months. -
Infection progressed from one lineage to coinfection with a second lineage, before clearance of coinfection and reinfection with a third, different lineage. -
Coinfection was difficult to identify through genomic methods. Published in Virology Journal (Jan. 4, 2024): https://doi.org/10.1186/s12985-023-02278-7
|
|
Scooped by
Juan Lama
|
In a recent study posted to the bioRxiv* preprint server, researchers analyzed the viral genomes of 28 endemic viruses to study the evolution of the ability of viruses to evade the neutralizing antibodies elicited by vaccines or previous infections. Background Viruses evolve rapidly and adapt to changing environments due to their high mutation rates and low generation time. Often viruses adapted to different animal hosts infect humans and optimize the methods through which they enter and replicate in the host cell, increasing the human-to-human transmission and evolving into a novel pathogen. The early stages of pandemics are often characterized by high adaptive evolutionary rates, as was seen during the coronavirus disease 2019 (COVID-19) pandemic and outbreaks related to various other respiratory viruses. While some viruses become endemic after adapting to a new host and do not evolve further, other endemic viruses continue to adapt through antigenic evolution, resulting in an arms race between the virus and the human immune system. Since viruses that undergo antigenic evolution pose the risk of repeat infections and increase their ability to evade vaccine-induced immunity, understanding which viruses continue to undergo antigen evolution could help manage future disease outbreaks. About the study The present study used sequence data for each gene in 28 viral genomes to estimate adaptive evolutionary rates. These 28 viruses spanned ten families and were transmitted between humans through various modes. Viruses with potentially high antigenic evolution rates were identified based on the high evolutionary rates for the genes coding for receptor-binding proteins since the receptor-binding region is involved in antibody neutralization and harbors most mutations that allow antigenic escape. The adaptive evolutionary rates were calculated in terms of the number of adaptive mutations in each codon per year, which allowed the adaptive evolutionary rates to be compared across the various genes in the genome and across viruses. The adaptive evolutionary rates of three viruses that were known to undergo antigenic evolution — coronavirus 229E, influenza viruses A/H3N2, and influenza viruses B/Yam — were compared against the evolutionary rates of three antigenically stable viruses, hepatitis A, measles, and influenza C/Yamagata. To understand the patterns of adaptive evolution in recent years, the sequence data for 28 viruses that are pathogenic to humans were obtained and curated. These viruses included deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) viruses and were transmitted between humans through bodily fluids, blood, vectors, fecal-oral, and respiratory routes. The researchers only investigated endemic viruses since they were interested in understanding the antigenic evolution that occurs during the endemic phase and not the initial adaptive phase. The evolutionary rates of the receptor binding protein, which was expected to be highly variable across endemic viruses, and the polymerase gene, which was expected to be conserved, were compared across the 28 viruses. Since the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been relatively recent, and the Omicron variant carried a large number of mutations indicating a single fixation event, SARS-CoV-2 was compared with ten other antigenically evolving viruses by comparing the amino-acid substitution rates in the receptor binding protein. Results The results reported that 10 of the 28 viruses undergo adaptive evolution resulting in the antigenic mutations that allow the viruses to escape the immunity induced by previous infections and vaccines. Furthermore, comparing amino-acid substitution rates between SARS-CoV-2 and other viruses revealed that SARS-CoV-2 is evolving and accumulating mutations that cause protein-coding changes at rates much higher than other endemic viruses. Antigenic evolution was found to be more prevalent in RNA viruses. Still, the researchers believe that since the list of viruses included in the study was not comprehensive and comprised only well-studied viruses, determining the proportion of antigenically evolving endemic viruses is difficult. Furthermore, the rate of adaptive mutations might not reflect the phenotypic changes occurring in the viruses, implying that viruses with receptor-binding protein genes that evolve at the same rates might not develop the ability to evade vaccine or infection-induced immunity at the same rates. Conclusions Overall, the findings suggested that many human viruses that have become endemic continue to evolve antigenically, gaining the ability to escape the neutralizing antibodies generated by vaccinations or previous infections. Ten of the 28 viruses investigated in this study showed continued adaptive evolution. In contrast, the amino-acid substitution rates showed that SARS-CoV-2 is evolving faster than the other ten endemic human viruses. Cited ressearch available in bioRxiv (May 22, 2023): https://doi.org/10.1101/2023.05.19.541367
|
|
Scooped by
Juan Lama
|
A study employing CRISPR/Cas9 to explore the evolutionary beginnings of some giant viruses finds evidence their large genomes arose from gene duplications. Most viruses are small and carry minimalist genomes. Even one of the largest small viruses, Vaccinia, measures merely one-fiftieth the size of a pollen grain and contains only 270 genes. Giant viruses flout these rules. With sizes that rival small bacteria and genomes that contain thousands of genes, their complexity emulates that of cellular life. How these viruses came to be so large has been the subject of much debate. Now, scientists are finally poised to unravel the mystery of their evolutionary origins, thanks to a suite of CRISPR/Cas9-based tools described in a Nature Communications paper from January. “It was by chance that we encountered the first giant virus,” says Chantal Abergel, a virologist at Aix-Marseille University in France. “It was Mimivirus, and it was actually mistaken for a bacterium.” In the 20 years since that discovery, virologists have prioritized exploring the diversity of giant viruses. Now that they’ve found a fair few, the focus has shifted towards studying their evolution in more detail with molecular biology techniques. Evolutionary biologists have grappled over two possible origins of giant viruses. One possibility is that they were once cellular organisms that shrunk physically and genetically over time. But most virologists now suspect giant viruses grew out of much smaller ones—though the evidence supporting either hypothesis is scant. To begin addressing this origin question, Abergel decided to examine how the essential genes in the Pandoravirus genome are distributed. In cellular organisms, essential genes are scattered throughout the genome—so if giant viruses are essentially reduced cells, one would expect a similar pattern. Alternatively, if the genes are clumped, that could indicate the viruses’ large genomes started out in a more compact form. One way to locate a virus’s essential genes is to knock out genes one at a time to find the ones that are needed for virus production. But to do that with a giant virus, Abergel needed a gene-editing system that worked in members of the group. With the help of Hugo Bisio, a postdoctoral researcher in Abergel’s lab, and colleagues at Aix–Marseille University, Abergel used a CRISPR/Cas9-based gene-editing system to modify the genome of the amoeba Acanthamoeba castellanii and the giant virus Pandoravirus neocaledonia, which infects it. The CRISPR/Cas9 system was designed to delete specific genes and consists of two guide RNAs and a Cas9 scission enzyme. Similar to other CRISPR/Cas9 systems, each guide RNA contains 17 to 20 bases designed to bind to one specific location on the genome of the giant virus or the amoeba, allowing the Cas9 scission enzyme to cut the genome at that site. The amoeba A. castellanii contains 25 copies of each chromosome, making it difficult to design an efficient CRISPR/Cas9 system that could delete each gene copy. To overcome this issue, the researchers modified their CRISPR/Cas9 system to generate a chain reaction. Each time DNA was cut to remove a gene, a DNA segment encoding the Cas9 enzyme and the guide RNAs responsible for the cut would take the place of the missing gene in the genome. This allowed gene deletions to repeat and propagate until all copies were removed. Once they optimized their CRISPR/Cas9 system, the team deleted each gene separately from the Pandoravirus genome and measured the resulting change in virus production, in order to determine how important each gene is to the virus’s lifecycle. They found that essential genes clustered together at one end of the genome and were segregated from nonessential genes at the other end. This level of gene orderliness has not been seen in viruses, according to Bisio. Even bacterial genomes aren’t quite so tidy: While they do group genes with linked functions together into gene clusters known as operons, these tend to be dispersed throughout the genome rather than grouped all together in one spot. Bisio says the cluster of essential genes may echo a smaller “core genome” of an ancient virus. This genome could have become elongated through multiple rounds of gene duplication that were biased in one direction to produce an additional set of spare nonessential genes. This could explain how modern-day giant viruses came to possess thousands of genes. “Our data indicate that complex viruses arose from smaller and simpler ones,” Bisio tells The Scientist in an email—noting that it will take further research to determine whether that’s true of all giant viruses or just Pandoravirus. Other studies found that some genes in giant viruses were usurped from their amoeba hosts, suggesting gene exchange is another way giant viruses increased in size. The team then set their sights on one of the many evolutionary mysteries of Pandoravirus: its lack of a capsid. Small viruses package their genomes into capsids made of viral proteins. While some giant viruses, such as Mimivirus, continue this tradition, others, including Pandoravirus, do not. If giant viruses did indeed evolve from smaller ones, there could be traces of capsid proteins hiding in their genomes. So, the researchers set out to study the function of potential capsid protein remnants in a close cousin of Pandoravirus, the smaller Mollivirus, which can also infect A. castellanii. Researchers have suspected that a Mollivirus protein called ml_347 evolved from a capsid gene based on its gene’s sequence and predicted 3D shape. So, the team investigated its function by deleting the gene using their CRISPR/Cas9 system. They found that the gene is important for Mollivirus assembly, which the authors say is intriguing given its possible capsid ancestry. It’s possible that, as capsids were lost in giant virus evolution, obsolete capsid genes were adapted for new assembly functions. Frederik Schulz, an evolutionary biologist with the DOE Joint Genome Institute in California who wasn’t involved with the study but who has worked with Chantal Abergel in the past, tells The Scientist that the findings align with recent discoveries. “There was a debate for a long time [about] how giant virus[es] evolved,” he says. “The working hypothesis in the previous years was that they evolved from smaller viruses, and that’s exactly what Chantal and her team could show and confirm using their CRISPR/Cas9 gene-editing approach.” Schulz notes that it will be exciting to see the CRISPR/Cas9 technology introduced into other host species, such as algae, which would allow researchers to expand research into a greater variety of giant viruses. He also points out that the system only works for viruses that replicate in a host cell’s nucleus, while most giant viruses replicate in cytoplasmic structures called viral factories, which the Cas9 enzyme and guide RNAs can’t penetrate. Still, Bisio says there’s much left to discover in Pandoravirus. “[This CRISPR/Cas9 technology is] a goldmine to find new functions,” he says—one that he and his colleagues are eager to employ to tease apart what all the virus’s genes do. Cited research published in Nat. Communications (Jan. 26, 2023): https://doi.org/10.1038/s41467-023-36145-4
|
|
Scooped by
Juan Lama
|
The milder clinical manifestations of Omicron infection relative to pre-Omicron SARS-CoV-2 raises the possibility that extensive evolution results in reduced pathogenicity. To test this hypothesis, we quantified induction of cell fusion and cell death in SARS-CoV-2 evolved from ancestral virus during long-term infection. Both cell fusion and death were reduced in Omicron BA.1 infection relative to ancestral virus. Evolved virus was isolated at different times during a 6-month infection in an immunosuppressed individual with advanced HIV disease. The virus isolated 16 days post-reported symptom onset induced fusogenicity and cell death at levels similar to BA.1. However, fusogenicity was increased in virus isolated at 6 months post-symptoms to levels intermediate between BA.1 and ancestral SARS-CoV-2. Similarly, infected cell death showed a graded increase from earlier to later isolates. These results may indicate that, at least by the cellular measures used here, evolution in long-term infection does not necessarily attenuate the virus. Preprint available at medRxiv (Nov. 24, 2022): https://doi.org/10.1101/2022.11.23.22282673
|
|
Scooped by
Juan Lama
|
This phylogenetic tree represents the clade distribution of global SARS-CoV-2 viral genomes since the beginning of the pandemic. In the center is the reference Wuhan-1 strain, with outside samples now separated by nucleotide divergence. The last few months have led to an explosion of new variants with further mutations in the viral spike resulting in further antibody evasion, and also carrying changes in many other viral proteins. Some recently sequenced viral genomes from samples collected in Southeast Asia (not shown in this figure) display over a hundred nucleotides changes when compared to Wuhan-1. The graph was generated with nextstrain.org
|
|
Scooped by
Juan Lama
|
Nature is analog. It is not a binary system. In the living world there are no explicit switches that discreetly turn systems on or off. Rather, nature adjusts systems through analog dials, like an old radio — gradually changing variables to achieve balance and equilibrium to ensure that life is sustainable and carries on. Evolution proceeds in this way, with new life forms appearing and some disappearing over millennia — or, in the case of microbial pathogens (viruses, bacteria and parasites) over days or weeks. Evolutionary change results from two opposing forces: Positive selection reproduces beneficial genetic variations that enable the virus to survive, while negative selection pressure hinders the virus’s survival and ability to reproduce. Evolution can be studied at the molecular level. For many years, my research was focused on the African trypanosome, the parasite responsible for African sleeping sickness. Antigenic variation Trypanosomes live in the bloodstream of its mammalian hosts (including humans) and early observations of their numbers showed a consistent wave-like pattern of increases followed by declining numbers and then, after a week or so, rising numbers again. Trypanosomes are vulnerable to the antibodies produced by their host’s immune system, which bind to the parasite and eliminate it. This immune response causes the trypanosome numbers to drop, as illustrated by the low points of the wave pattern. But before the trypanosomes disappear entirely, their numbers rise again and the wave repeats. This intriguing growth pattern generated much interest and research in my laboratory and, ultimately, we learned that the parasite can alter its molecular identity to evade the host’s antibodies before it is completely eliminated. This means that the population of trypanosomes responsible for each of the wave peaks is a variant distinct from all the others. Antibodies directed against one variant have no effect on subsequent variants, so the wave pattern continues. The trypanosome’s very successful strategy evolved to help it survive in the face of constant negative selection pressure from antibodies. This mechanism that helps a parasite or pathogen evade the host’s immune system is called antigenic variation. COVID-19’s waves are similar to sleeping sickness I am reminded of the growth curve of trypanosomes when looking at the pattern of Canadian case counts from the ongoing COVID-19 pandemic. The peaks in cases reflect the arrival of new variants, the most recent of which is omicron, the variant now circulating most widely globally. The strategy used by SARS-CoV-2, the virus that causes COVID-19, is similar to the trypanosome’s, although the mechanism for generating novel variants is quite different. For the virus, new variants arise by mutation in genes that encode the so-called “spike protein,” the part of the virus that enables it to enter cells and infect people. Mutations arise due to “errors” that occur when the virus is replicating itself in the cells of the host’s respiratory system. Because the virus has a mechanism that can attempt to repair the “errors,” SARS-CoV-2 evolves more slowly than the trypanosome. It evolves more slowly because the virus has a mechanism that can try to repair the “errors.” However, this repair process is not perfect, and some mutations get retained. If mutations result in a spike protein distinct from any other variant preceding it, we will see a new variant appearing. The omicron variant is particularly interesting (and somewhat ominous) because of its high number of mutations, not only in the spike protein but in other viral genes as well. By employing this strategy of antigenic variation, the survival of the SARS-CoV-2 virus is assured. So, the appearance of new variants is due to mutations that represent the positive selection force: genetic variations that help the organism get reproduced. The decline of case numbers during a pandemic is due to negative selection forces. These include effective public health interventions that limit the spread from one person to the next (such as masks), as well as the hosts’ immune response (antibodies) resulting from either infection, vaccination or both. An infected person will, over time, generate antibodies against the virus and begin to eliminate that variant, like in the trypanosome case. But because SARS-CoV-2 mutations occur slowly, the virus needs to find a new, non-immune person to carry on. In order to find new non-immune hosts, the virus induces symptoms that help it to spread: the coughing and sneezing that enable it to jump from one person to the next via droplets. Antibodies and illness Given the capacity of SARS-CoV-2 to mutate, there are certainly new variants arising continuously. However, if medical and public health interventions are successful in reducing transmission between infected and uninfected/unvaccinated people, it is quite possible that the virus will evolve to generate a less virulent variant that could establish itself as an endemic infection producing mild symptoms. When people infected with a pathogenic microbe experience symptoms of illness, those symptoms often serve a purpose: they can contribute to either the microbe’s survival or the survival of the infected host. A classic case is diarrhea resulting from infection with cholera or from amoebic dysentery. Both infections produce life-threatening diarrhea, but the symptom serves different purposes in each disease. In the case of cholera, this symptom serves the microbe because it enables the bacteria to exit the host’s body and, in places with poor sanitation, contaminate the water supply and transmit to new hosts. In the case of amoebic dysentery, the symptom is a result of the host’s body attempting to rid itself of the infection. Clinicians must be able to distinguish between these two scenarios in the management of infectious diseases in order to avoid contributing to the problem rather than solving it. In the case of COVID-19, clinical symptoms like sneezing and coughing that enable the virus to spread through the air are positively selecting variants that help the virus spread to new, susceptible individuals (such as unvaccinated people). That means measures like masking, social distancing, and vaccination can impede spread by helping to prevent aerosol transmission. Continued efforts to achieve a fully vaccinated population are crucial. The unvaccinated and the uninfected are ideal hosts for SARS-CoV-2, and ideal for generating new variants due to the absence of negative selection by antibodies, which makes it easier for the virus to replicate and produce new mutations. Although nature may move slowly in an analog manner, humans can flip binary switches and we can act now to ensure global vaccine equity. Ensuring global vaccine coverage is not only imperative from an evolutionary perspective but is clearly the ethical option as well. Written by Michael Clarke, Adjunct Professor, Interfaculty Program in Public Health, Schulich School of Medicine and Dentistry, Western University. This article was first published in The Conversation.
|
|
Scooped by
Juan Lama
|
Souilmi et al. find that strong genetic adaptation occurred in human East Asian populations, at multiple genes that interact with coronaviruses, including SARS-CoV-2. The adaptation
started 25,000 years ago, and functional analysis of the adapting genes supports the occurrence of a corona- or related virus epidemic around that time in East Asia. Highlights - Ancient viral epidemics can be identified through adaptation in host genomes
- Genomes in East Asia bear the signature of an ∼25,000-year-old viral epidemic
- Functional analysis supports an ancient corona- or related virus epidemic
Summary The current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has emphasized the vulnerability of human populations to novel viral pressures, despite the vast array of epidemiological and biomedical tools now available. Notably, modern human genomes contain evolutionary information tracing back tens of thousands of years, which may help identify the viruses that have impacted our ancestors—pointing to which viruses have future pandemic potential. Here, we apply evolutionary analyses to human genomic datasets to recover selection events involving tens of human genes that interact with coronaviruses, including SARS-CoV-2, that likely started more than 20,000 years ago. These adaptive events were limited to the population ancestral to East Asian populations. Multiple lines of functional evidence support an ancient viral selective pressure, and East Asia is the geographical origin of several modern coronavirus epidemics. An arms race with an ancient coronavirus, or with a different virus that happened to use similar interactions as coronaviruses with human hosts, may thus have taken place in ancestral East Asian populations. By learning more about our ancient viral foes, our study highlights the promise of evolutionary information to better predict the pandemics of the future. Importantly, adaptation to ancient viral epidemics in specific human populations does not necessarily imply any difference in genetic susceptibility between different human populations, and the current evidence points toward an overwhelming impact of socioeconomic factors in the case of coronavirus disease 2019 (COVID-19). Published in Current Biology (June 24, 2021): https://doi.org/10.1016/j.cub.2021.05.067
|
|
Scooped by
Juan Lama
|
Nucleotide substitution rate of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is relatively low compared to the other RNA viruses because coronaviruses including SARS-CoV-2 encode non-structural protein 14 (nsp14) that is an error-correcting exonuclease protein. In this study, to understand genome evolution of SARS-CoV-2 in the current pandemic, we examined mutations of SARS-CoV-2 nsp14 which could inhibit its error-correcting function. First, to obtain functionally important sites of nsp14, we examined 62 representative coronaviruses belonging to alpha, beta, gamma, delta, and unclassified coronaviruses. As a result, 99 out of 527 amino acid sites of nsp14 were evolutionarily conserved. We then examined nsp14 sequences obtained from 28,082 SARS-CoV-2 genomes and identified 6 amino acid changes in nsp14 mutants that were not detected in the 62 representative coronaviruses. We examined genome substitution rates of these mutants and found that an nsp14 mutant with a proline to leucine change at position 203 (P203L) showed a higher substitution rate (35.9 substitutions/year) than SARS-CoV-2 possessing wild-type nsp14 (19.8 substitutions/year). We confirmed that the substitution rate of the P203L is significantly higher than those of other variants containing mutations in structural proteins. Although the number of SARS-CoV-2 variants containing P203L mutation of nsp14 is limited (26), these mutants appeared at least 10 times independently in the current pandemic. These results indicated that the molecular function of nsp14 is important for survival of various coronaviruses including SARS-CoV-2 and that some mutations such as P203L of nsp14 inhibiting its error-correcting function are removed rapidly due to their deleterious effects. Preprint available in bioRxiv (Dec. 26, 2020): https://doi.org/10.1101/2020.12.23.424231
|
|
Scooped by
Juan Lama
|
The new coronavirus resurged again and again in the body of an infected man, eventually killing him while showing evidence of fast-paced evolution. Manuela Cernadas and Jonathan Li at Brigham and Women’s Hospital in Boston, Massachusetts, and their colleagues followed the course of COVID-19 in a 45-year-old man with a long-standing autoimmune disorder, who was on a medication regimen that included powerful immunosuppressants (B. Choi et al. N. Engl. J. Med. https://doi.org/fhv8; 2020). Roughly 40 days after the man first tested positive for SARS-CoV-2, follow-up tests indicated that the virus was dwindling — but it surged back, despite antiviral treatment. The man’s infection subsided and then returned twice more before he died, five months after his first COVID-19 diagnosis. Genomic analysis showed that the man had not been infected multiple times. Instead, the virus had lingered and quickly mutated in his body. Original study published in NEJM (Nov. 11, 2020): https://doi.org/10.1056/NEJMc2031364
|
|
Scooped by
Juan Lama
|
A team of scientists studying the origin of SARS-CoV-2, the virus that has caused the COVID-19 pandemic, found that it was especially well-suited to jump from animals to humans by shapeshifting as it gained the ability to infect human cells. Conducting a genetic analysis, researchers from Duke University, Los Alamos National Laboratory, the University of Texas at El Paso and New York University confirmed that the closest relative of the virus was a coronavirus that infects bats. But that virus's ability to infect humans was gained through exchanging a critical gene fragment from a coronavirus that infects a scaly mammal called a pangolin, which made it possible for the virus to infect humans. The researchers report that this jump from species-to-species was the result of the virus's ability to bind to host cells through alterations in its genetic material. By analogy, it is as if the virus retooled the key that enables it to unlock a host cell's door -- in this case a human cell. In the case of SARS-CoV-2, the "key" is a spike protein found on the surface of the virus. Coronaviruses use this protein to attach to cells and infect them. "Very much like the original SARS that jumped from bats to civets, or MERS that went from bats to dromedary camels, and then to humans, the progenitor of this pandemic coronavirus underwent evolutionary changes in its genetic material that enabled it to eventually infect humans," said Feng Gao, M.D., professor of medicine in the Division of Infectious Diseases at Duke University School of Medicine and corresponding author of the study publishing online May 29 in the journal Science Advances. The researchers found that typical pangolin coronaviruses are too different from SARS-CoV-2 for them to have directly caused the human pandemic. However, they do contain a receptor-binding site -- a part of the spike protein necessary to bind to the cell membrane -- that is important for human infection. This binding site makes it possible to affix to a cell surface protein that is abundant on human respiratory and intestinal epithelial cells, endothelial cell and kidney cells, among others. While the viral ancestor in the bat is the most closely related coronavirus to SARS-CoV-2, its binding site is very different, and on its own cannot efficiently infect human cells. SARS-CoV-2 appears to be a hybrid between bat and pangolin viruses to obtain the "key" necessary receptor-binding site for human infection. Original study published in Science Advances (May 29, 2020): https://doi.org/10.1126/sciadv.abb9153
|
|
Scooped by
Juan Lama
|
The second notable feature of SARS-CoV-2 is a predicted polybasic cleavage site (RRAR) in the spike protein at the junction of S1 and S2, the two subunits of the spike protein (Figure 1b)8,9. In addition to two basic arginines and an alanine at the cleavage site, a leading proline is also inserted; thus, the fully inserted sequence is PRRA (Figure 1b). The strong turn created by the proline insertion is predicted to result in the addition of O-linked glycans to S673, T678, and S686 that flank the polybasic cleavage site. A polybasic cleavage site has not previously been observed in related lineage B betacoronaviruses and is a unique feature of SARS-CoV-2. Some human betacoronaviruses, including HCoV-HKU1 (lineage A), have polybasic cleavage sites, as well as predicted O-linked glycans near the S1/S2 cleavage site. While the functional consequence of the polybasic cleavage site in SARS-CoV-2 is unknown, experiments with SARS-CoV have shown that engineering such a site at the S1/S2 junction enhances cell–cell fusion but does not affect virus entry10. Polybasic cleavage sites allow effective cleavage by furin and other proteases, and can be acquired at the junction of the two subunits of the haemagglutinin (HA) protein of avian influenza viruses in conditions that select for rapid virus replication and transmission (e.g. highly dense chicken populations). HA serves a similar function in cell-cell fusion and viral entry as the coronavirus S protein. Acquisition of a polybasic cleavage site in HA, by either insertion or recombination, converts low pathogenicity avian influenza viruses into highly pathogenic forms11-13. The acquisition of polybasic cleavage sites by the influenza virus HA has also been observed after repeated forced passage in cell culture or through animals14,15. Similarly, an avirulent isolate of Newcastle Disease virus became highly pathogenic during serial passage in chickens by incremental acquisition of a polybasic cleavage site at the junction of its fusion protein subunits.... ...It is improbable that SARS-CoV-2 emerged through laboratory manipulation of an existing SARS-related coronavirus. As noted above, the RBD of SARS-CoV-2 is optimized for human ACE2 receptor binding with an efficient binding solution different to that which would have been predicted. Further, if genetic manipulation had been performed, one would expect that one of the several reverse genetic systems available for betacoronaviruses would have been used. However, this is not the case as the genetic data shows that SARS-CoV-2 is not derived from any previously used virus backbone17. Instead, we propose two scenarios that can plausibly explain the origin of SARS-CoV-2: (i) natural selection in a non-human animal host prior to zoonotic transfer, and (ii) natural selection in humans following zoonotic transfer. We also discuss whether selection during passage in culture could have given rise to the same observed features... Mandarin version of the article available at: http://virological.org/uploads/short-url/dHpDmgjWKLNlBztUWteEJZq4Pvw.pdf
|