NCBI Gene (formerly Entrez) lets you access the wealth of information at NCBI.
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account


 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
NCBI Gene (formerly Entrez) lets you access the wealth of information at NCBI. 
No comment yet.
Sign up to comment

Detection of regions of genomic sequences that are rich in the CpG pattern is important because such regions are resistant to methylation and tend to be associated with genes which are frequently switched on. EBI's EMBOSS can be used to find CpG islands in a nucleotide sequence.
OTHER SITES:
Pepinfo (EMBOSS)

AUGUSTUS is a database for ab initio prediction of alternative transcripts. You can predict a gene in eukaryotic genomic sequences based on a generalized hidden Markov model.
|
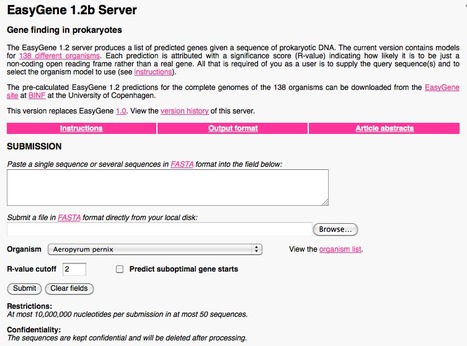
EasyGene is a prokaryotic gene finder that ranks ORFs by statistical significance. Predict and statistically evaluate predicted prokaryotic open reading frames (ORFs).
Dr. Stefan Gruenwald's insight:
Complexity is a web resource for the analysis of DNA sequence complexity. Search for low complexity regions in long sequences and estimate complexity of groups of aligned sequences.

BLogo is a tool for the visualization of a bias in biological sequences. It detects and displays statistically significant position-specific sequence bias with reduced background noise.

AGenDA is a gene-prediction tool that is based on cross-species sequence comparison. The software tools CHAOS and DIALIGN are used to align a pair of syntenic genomic sequences, e.g. from human and mouse. AGenDA searches for conserved splice sites and start/stop codons at the boundaries of identified sequence homologies in order to identify potential exons; these potential exons are then used to assemble entire gene models.
|