


Mayo Clinic scientists have developed an immunotherapy strategy that potentially lays the groundwork for treating a spectrum of autoimmune diseases.
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account


 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
Mayo Clinic scientists have developed an immunotherapy strategy that potentially lays the groundwork for treating a spectrum of autoimmune diseases. 
No comment yet.
Sign up to comment
At EPFL's School of Engineering, Professor Li Tang's Laboratory of Biomaterials for Immunoengineering has made significant strides in cancer treatment research.
BigField GEG Tech's insight:
Traditional CAR-T cells, while effective against liquid cancers, face challenges in solid tumors: the cells wear out and ultimately fail to destroy the cancer completely. Ground-breaking research is providing an innovative approach to this challenge. Researchers are introducing CAR-T cells that excrete the IL-10 molecule. In other words, the cell has been designed to produce its own "drug" to stay healthy in the tumor's hostile environment. In the laboratory, this innovative CAR-T therapy systematically eradicated cancerous tumors in mouse models. What's more, in ongoing clinical trials, eleven patients have appeared to achieve complete remission with this treatment, representing a 100% success rate to date. Notably, the evidence from the laboratory study suggests the long-term efficacy of the therapy, and indicates that its manufacture could be both faster and more cost-effective than current methods
In the context of relapsed and refractory childhood pre-B cell acute lymphoblastic leukemia (R/R B-ALL), CD19-targeting chimeric antigen receptor (CAR)-T cells often induce durable remissions, which requires the persistence of CAR-T cells. In this study, we systematically analyzed CD19 CAR-T cells of 10 children with R/R B-ALL enrolled in the CARPALL trial via high-throughput single-cell gene expression and T cell receptor sequencing of infusion products and serial blood and bone marrow samples up to 5 years after infusion. We show that long-lived CAR-T cells developed a CD4/CD8 double-negative phenotype with an exhausted-like memory state and distinct transcriptional signature. This persistence signature was dominant among circulating CAR-T cells in all children with a long-lived treatment response for which sequencing data were sufficient (4/4, 100%). The signature was also present across T cell subsets and clonotypes, indicating that persisting CAR-T cells converge transcriptionally. This persistence signature was also detected in two adult patients with chronic lymphocytic leukemia with decade-long remissions who received a different CD19 CAR-T cell product. Examination of single T cell transcriptomes from a wide range of healthy and diseased tissues across children and adults indicated that the persistence signature may be specific to long-lived CAR-T cells. These findings raise the possibility that a universal transcriptional signature of clinically effective, persistent CD19 CAR-T cells exists. In children with relapsed or refractory B cell acute lymphoblastic leukemia and in complete remission after CD19 CAR-T cell therapy, long-lived CAR-T cells express a persistence gene signature that is also present in persistent CD19 CAR-T cells from adults with chronic lymphocytic leukemia.
BigField GEG Tech's insight:
CAR T cells have become an established treatment option for children with a rare form of relapsed or incurable leukemia. One of the key factors determining whether treatment will lead to lasting remission of leukemia is how long the CAR T cells live in the body. One team was able to study the cells of 10 children enrolled in a pioneering clinical trial (CARPALL) for up to five years after their initial CAR T cell treatment. This has enabled them to better understand why some of these CAR T cells remain in a patient's bloodstream, and why others disappear early, potentially allowing the cancer to recur. Using techniques that analyze individual cells at the genetic level to understand what they do, the scientists were able to identify a unique "signature" in long-lived CAR T cells. The signature suggested that long-lived CAR T cells in the blood transformed into a different state that allowed them to continue monitoring the patient's body for cancer cells. As part of the study, the researchers identified key genes in CAR T cells that appeared to enable them to persist in the body for a long time. These genes will provide a starting point for future studies to identify markers of persistence in CAR T-cell products as they are manufactured, and ultimately to improve their efficacy.
Adoptive transfer of genetically engineered chimeric antigen receptor (CAR) T cells is becoming a promising treatment option for hematological malignancies. However, T cell immunotherapies have mostly failed in individuals with solid tumors. Here, with a CRISPR–Cas9 pooled library, we performed an in vivo targeted loss-of-function screen and identified ST3 β-galactoside α-2,3-sialyltransferase 1 (ST3GAL1) as a negative regulator of the cancer-specific migration of CAR T cells. Analysis of glycosylated proteins revealed that CD18 is a major effector of ST3GAL1 in activated CD8+ T cells. ST3GAL1-mediated glycosylation induces the spontaneous nonspecific tissue sequestration of T cells by altering lymphocyte function-associated antigen-1 (LFA-1) endocytic recycling. Engineered CAR T cells with enhanced expression of βII-spectrin, a central LFA-1-associated cytoskeleton molecule, reversed ST3GAL1-mediated nonspecific T cell migration and reduced tumor growth in mice by improving tumor-specific homing of CAR T cells. These findings identify the ST3GAL1–βII-spectrin axis as a major cell-intrinsic program for cancer-targeting CAR T cell migration and as a promising strategy for effective T cell immunotherapy. CAR T cell success requires targeting tumors, but these cells can get trapped in other tissues, such as in the lungs, where they can cause pathology. Here, the authors use a loss-of-function CRISPR screen to identify regulators of CAR T cell tumor trafficking and engineer CAR T cells accordingly to overcome this limitation.
BigField GEG Tech's insight:
Immunotherapy, particularly CAR T-Cell cancer therapy, extends the lives of many patients. But sometimes the therapy randomly migrates to places it shouldn't go, sneaking into the lungs or other non-cancerous tissue and causing toxic side effects. However, a team of researchers has discovered the molecule responsible for guiding T cells to tumors, setting the stage for scientists to improve the revolutionary treatment. Their discovery of the crucial migration control gene that expresses ST3GAL1 is the result of "unbiased genomic screening": researchers used a state-of-the-art CRISPR technique to edit thousands of genes expressed in T cells, then tested the migration control capabilities of these genes, one by one over a period of nearly four years, in mouse models. The next step is to find a drug that can manipulate the key T cell protein, ST3GAL1. If the study progresses as planned, such a drug could be added to the CAR T-cell regimen to ensure that the T cells reach their targets

With a slew of tools to trick out immune cells, researchers are expanding the repertoire of CAR-T therapies.
BigField GEG Tech's insight:
Crystal Mackall remembers her scepticism the first time she heard a talk about a way to engineer T cells to recognize and kill cancer. Sitting in the audience at a 1996 meeting in Germany, the paediatric oncologist turned to the person next to her and said: “No way. That’s too crazy.” Today, things are different. “I’ve been humbled,” says Mackall, who now works at Stanford University in California developing such cells to treat brain tumours.
Scientists at St. Jude Children's Research Hospital identified a molecular mechanism that in a preclinical study unlocked the promise of CAR T–cell therapy for treatment of solid tumors.
BigField GEG Tech's insight:
Currently, too few CAR T cells become memory cells that persist and create more T cells in the long term. However, a group of researchers recently showed that the distribution of the c-Myc protein in a parental T cell may be important for this process and published this work in the journal Nature. The researchers knew that a daughter cell with more c-Myc became an effector cell. In this study, the team found that the protein complex cBAF (canonical Brg1/Brg-associated factor) interacted with c-Myc. Daughter cells with high concentrations of cBAF and c-Myc became effector T cells. The cBAF binds certain regions of chromatin, proteins on DNA. The discovery suggests that it can guide the fate of cells, what type of T cells they become, by controlling the expression of effector cell-related genes. The distribution of cBAF occurs in the first activated T cell that begins the adaptive immune response; therefore, the researchers realized that cell fate is decided early in the immune response. The researchers used the molecular information they discovered. They applied a cBAF inhibitor during CAR T cell activation to generate more memory T cells. In a preclinical model, T cells treated with an inhibitor-controlled tumor growth better than untreated cells. The treated cells also survived longer and in greater numbers.

Florencer Edwine's curator insight,
July 13, 2022 1:15 PM
https://comprarmorfina.com/ https://acquistaossicodone.eu/ acquista oxycontin achete de MORPHINE https://achetmorphine.com/ acquista Subutex Online https://acquistasubutex.com/
Genetically engineered immune cells target cancer cells that may be responsible for leukemia relapse
Genetically engineered immune cells successfully target the specific cancer cells that may be responsible for relapse of acute myeloid leukemia (AML), a type of blood cancer, and proved effective in animal models of the disease, according to a preclinical study by investigators at Weill Cornell Medicine.
BigField GEG Tech's insight:
Genetically modified immune cells successfully target specific cancer cells that may be responsible for the relapse of acute myeloid leukemia (AML). In a study published on 28 April in Nature Communications, the researchers developed a CAR T cell therapy (UCART123) targeting CD123, which is found on leukemia stem cells and enables T cells to seek out and attack cancer cells. When the team tested the UCART123 cells in a mouse model of AML, they found that the therapy effectively eliminated leukemia cells and prolonged survival. The scientists also devised a highly sensitive monitoring strategy to detect any residual cancer cells and assess the persistence of UCART123 cells. Finally, they demonstrated that UCART123 cells have specificity against leukemia cells, with minimal toxicity to normal blood cells in mice. The preclinical results led to a Phase 1 clinical trial testing UCART123 in patients with relapsed/refractory AML at several sites across the US, including New York-Presbyterian/Weill Cornell Medical Center. The results of the preclinical study suggest that UCART123 cells are highly selective and specific in targeting AML.
In recent years, great advances have been made in the development of new successful immunotherapies to treat cancer. CAR T-cell therapy and antibody treatments are two types of targeted immunotherapies that have revolutionized areas of cancer care.
BigField GEG Tech's insight:
CAR T-cell therapy and antibody treatments are two types of targeted immunotherapies that have revolutionized the fields of cancer care. However, there are still significant challenges in identifying cancer cell surface proteins as targets for immunotherapies. A research group at Lund University in Sweden is well on their way as they have developed a new precision medicine technology that allows for comprehensive mapping of the entire tumor cell surface antigen landscape in patients. The method developed by the research team, "Tumor Surfaceome Mapping, TS-MAP," allows direct analysis of all accessible tumor cell surface antigens in patient tumor tissue. In a close collaboration between neurosurgery, oncology and advanced proteomics in Lund, the researchers were able to identify several tumor cell surface antigens in fresh tissue from patients with aggressive brain tumors for which there is currently no effective treatment. An important advantage of the TS-MAP technology is that it provides a complete picture of the cell surface antigens displayed on the surface of the cancer cell, as well as information about specific cell surface antigens that have a high capacity to infiltrate cancer cells, and can destroy them from within.
Clinicians who treat patients with chimeric antigen receptor T cells have become adept at identifying and treating acute neurotoxicity, a common adverse event associated with the therapy.Researchers from Mount Sinai published a case study in Nature Medicine about a patient who developed neurocognitive and hypokinetic movement disorder with features of Parkinson’s disease after receiving
BigField GEG Tech's insight:
Clinicians treating patients with CAR T cells have become adept at identifying and treating acute neurotoxicity, a common adverse event associated with the therapy. Researchers at Mount Sinai published a case study in Nature Medicine of a patient who developed a neurocognitive, hypokinetic movement disorder with features of Parkinson's disease after receiving the CAR-T therapy targeting the B-cell maturation antigen (BCMA) called ciltacabtagene autoleucel in the CARTITUDE-1 clinical trial. Since reports of this toxicity have not been observed with CD19-directed CAR-T therapies for other blood cancers, it appears to be specifically associated with BCMA-directed CAR-T. As it turns out, Parekh and colleagues found that BCMA is expressed on certain brain cells, and they saw evidence that BCMA-targeted CAR-T cells entered the patient's cerebrospinal fluid and crossed the blood-brain barrier. With ciltacabtagene autoleucel poised to become the second BCMA-directed CAR-T to gain commercial approval later this year, clinicians should discuss the potential benefits and risks of the therapy with their patients. They also need to be vigilant about screening for late neurological toxicities until researchers can make improvements to the new CAR-T constructs to mitigate the risk.

Fibrosis affects millions of people with cardiac disease. We developed a therapeutic approach to generate transient antifibrotic chimeric antigen receptor (CAR) T cells in vivo by deliverin
BigField GEG Tech's insight:
The standard CAR T cell strategy would be problematic when directed against heart failure or other fibrotic diseases in humans. Fibroblasts have a normal and important function in the body, particularly in wound healing. CAR T cells that are genetically reprogrammed to attack fibroblasts could survive in the body for months or even years, suppressing the fibroblast population and impairing wound healing for all that time. Therefore, in the new study published in Science, Epstein and colleagues designed a technique for a more temporary and controllable, and much more procedurally simple, type of CAR T cell therapy. They designed an mRNA that encodes a T-cell receptor targeting activated fibroblasts and encapsulated the mRNA in tiny bubble-like lipid nanoparticles, which are themselves coated with molecules that lodge in T cells. Injections of this therapy into mice modeling heart failure successfully reprogrammed a large population of mouse T cells, causing a major reduction in cardiac fibrosis in the animals and restoration of a mostly normal heart size and function with no sign of continued anti-fibroblast T cell activity one week after treatment. Researchers continue to test this mRNA-based transient CAR T-cell technology, with the hope of eventually starting clinical trials
BigField GEG Tech's insight:
According to Jing Pan's article in the Journal of Clinical Oncology titled: Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human phase I trial, CAR T cells are reported to be remarkably effective in patients with B-cell acute lymphoblastic leukemia (ALL) but have not been successful to date in patients with T-cell ALL (T-ALL). Now, data from Pan and colleagues demonstrate the safety and impressive short-term efficacy of allogeneic donor-derived anti-CD7 CAR T cells in an early phase clinical trial involving patients with relapsed and/or refractory T-ALL.

Update on Allogene Therapeutics off-the-shelf CAR-T cancer therapies. This week’s gene-editing update looks at an investigational new drug (IND) programme and a pre-clinical programme for gene-edited CAR T-cell therapies for renal cell carcinoma, haematological cancers, and multiple myeloma.
BigField GEG Tech's insight:
Allogene Therapeutics develops allogeneic CAR T cell-based therapies for a range of hematological and solid cancers. Two candidates are being developed using Allogene's exclusive Allo CAR T platform :
BigField GEG Tech's insight:
Cellectis established a preclinical proof-of-concept for its new CAR T therapy in a study published in Nature Communications.
|
BigField GEG Tech's insight:
Researchers have reported promising results in a Phase I/II trial involving 37 patients with relapsed or refractory B-cell malignancies who were treated with a cord blood-derived natural killer (NK) chimeric antigen receptor (CAR), a cell therapy targeting CD19. Results showed an overall response (OR) rate of 48.6% 100 days after treatment, with one-year progression-free survival (PFS) and overall survival (OS) rates of 32% and 68%, respectively. The trial reported an excellent safety profile, with no cases of cytokine release syndrome (CRS), neurotoxicity or graft-versus-host disease. Another key finding of the trial was the importance of allogeneic cord blood donor selection criteria in the manufacture of CAR NK cells. Cord blood units cryopreserved within 24 hours of collection and those with a low content of nucleated red blood cells were associated with significantly better results. CAR NK cells generated from these units resulted in a one-year PFS rate of 69% and an OS rate of 94%, compared with 5% and 48%, respectively, for units with higher nucleated red cell content or longer collection to cryopreservation times.

Immunotherapy using modified chimeric antigen receptor (CAR) T cells has greatly improved survival rates for pediatric patients with relapsed and recurrent leukemia.
BigField GEG Tech's insight:
Solid tumors generate anti-immune signals that deactivate CAR T cells, making treatment less effective. To solve this problem, scientists have combined CAR T cells with cytokine injection, which can cause significant unintended toxicities. Researchers replaced the extracellular domain of various cytokine receptors with leucine zippers to create constitutively active receptors. CAR T cells expressing one of these chimeric cytokine receptors had superior antitumor activity against several types of cancer in cell lines and mouse models compared with conventional CAR T cells. Although chimeric cytokine receptors give a constant "on" signal to CAR T cells, they do not induce non-specific proliferation of CAR T cells. The system thus limits the effect of cytokine signaling to modified cells only, reduces the risk of cytokine-related toxicity, and provides a signal that these CAR T cells should function effectively in a suppressive tumor microenvironment.

runtzwraps's curator insight,
December 18, 2023 4:42 PM
https://runtzwrapz.com If you have any questions about our return coverage, please Really don't hesitate to Call us. We worth your business and take pleasure in the opportunity to serve you. Some are aged with strawberries to find the strawberry flavor, others are aged with vanilla, bourbon and cocoa to have the accurate vanilla taste we all appreciate. You may check out Agave that's aged with sweet honey and maple syrup. https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online/ https://runtzwrapz.com/product/buy-frosties-runtz-onlne/ https://runtzwrapz.com/product/buy-runtz-weed-online/ https://runtzwrapz.com/product/buy-sharklato-runtz/ https://runtzwrapz.com/product/buy-vanilla-cream-runtz-wraps/ https://runtzwrapz.com/product/cbd-gummy-bears-100mg/ https://runtzwrapz.com/product/ed-bills-lollipop-40mg/ https://runtzwrapz.com/product/buy-fresh-strawberry-runtz-wrap/ https://runtzwrapz.com/product/mango-kush-strain/ https://runtzwrapz.com/product/motorbreath-strain-near-me/ https://runtzwrapz.com/product/buy-natural-runtz-wraps-online/ https://runtzwrapz.com/product/white-runtz-strain-for-sale/ https://runtzwrapz.com If you have any questions about our return coverage, please Really don't hesitate to Call us. We worth your business and take pleasure in the opportunity to serve you. Some are aged with strawberries to find the strawberry flavor, others are aged with vanilla, bourbon and cocoa to have the accurate vanilla taste we all appreciate. You may check out Agave that's aged with sweet honey and maple syrup. https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online/ https://runtzwrapz.com/product/buy-frosties-runtz-onlne/ https://runtzwrapz.com/product/buy-runtz-weed-online/ https://runtzwrapz.com/product/buy-sharklato-runtz/ https://runtzwrapz.com/product/buy-vanilla-cream-runtz-wraps/ https://runtzwrapz.com/product/cbd-gummy-bears-100mg/ https://runtzwrapz.com/product/ed-bills-lollipop-40mg/ https://runtzwrapz.com/product/buy-fresh-strawberry-runtz-wrap/ https://runtzwrapz.com/product/mango-kush-strain/ https://runtzwrapz.com/product/motorbreath-strain-near-me/ https://runtzwrapz.com/product/buy-natural-runtz-wraps-online/ https://runtzwrapz.com/product/white-runtz-strain-for-sale/ https://runtzwrapz.com If you have any questions about our return coverage, please Really don't hesitate to Call us. We worth your business and take pleasure in the opportunity to serve you. Some are aged with strawberries to find the strawberry flavor, others are aged with vanilla, bourbon and cocoa to have the accurate vanilla taste we all appreciate. You may check out Agave that's aged with sweet honey and maple syrup. https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online https://runtzwrapz.com https://runtzwrapz.com/product/buy-banana-runtz-online/ https://runtzwrapz.com/product/big-runtz-strain-for-sale/ https://runtzwrapz.com/product/buy-agave-runtz-wrap-online/ https://runtzwrapz.com/product/buy-banana-split-runtz-wrap/ https://runtzwrapz.com/product/buy-berry-runtz/ https://runtzwrapz.com/product/buy-cake-mix-runtz-online/ https://runtzwrapz.com/product/buy-frosties-runtz-onlne/ https://runtzwrapz.com/product/buy-runtz-weed-online/ https://runtzwrapz.com/product/buy-sharklato-runtz/ https://runtzwrapz.com/product/buy-vanilla-cream-runtz-wraps/ https://runtzwrapz.com/product/cbd-gummy-bears-100mg/ https://runtzwrapz.com/product/ed-bills-lollipop-40mg/ https://runtzwrapz.com/product/buy-fresh-strawberry-runtz-wrap/ https://runtzwrapz.com/product/mango-kush-strain/ https://runtzwrapz.com/product/motorbreath-strain-near-me/ https://runtzwrapz.com/product/buy-natural-runtz-wraps-online/ https://runtzwrapz.com/product/white-runtz-strain-for-sale/

Chimeric antigen receptors (CARs) and synthetic Notch (synNotch) receptors are engineered cell-surface receptors that sense a target antigen and respond by activating T cell receptor signaling or a customized gene program, respectively. Here, to expand the targeting capabilities of these receptors, we develop “universal” receptor systems for which receptor specificity can be directed post-translationally via covalent attachment of a co-administered antibody bearing a benzylguanine (BG) motif. A SNAPtag self-labeling enzyme is genetically fused to the receptor and reacts with BG-conjugated antibodies for covalent assembly, programming antigen recognition. We demonstrate that activation of SNAP-CAR and SNAP-synNotch receptors can be successfully targeted by clinically relevant BG-conjugated antibodies, including anti-tumor activity of SNAP-CAR T cells in vivo in a human tumor xenograft mouse model. Finally, we develop a mathematical model to better define the parameters affecting universal receptor signaling. SNAP receptors provide a powerful strategy to post-translationally reprogram the targeting specificity of engineered cells. Chimeric antigen receptors (CARs) and synthetic Notch (synNotch) receptors are promising platforms for cell-based immunotherapies. Here, the authors develop highly programmable versions of these receptors that can be universally targeted to antigens of interest through covalent enzyme chemistry.
BigField GEG Tech's insight:
Researchers have developed a universal receptor system that allows T cells to recognize any cell surface target, enabling highly customizable CAR T cell and other immunotherapies for treating cancer and other diseases. The new approach involves engineering T cells with receptors bearing a universal "SNAPtag" that fuses with antibodies targeting different proteins. By tweaking the type or dose of these antibodies, treatments could be tailored for optimal immune responses. The researchers showed that their SNAP approach works in two important receptors: CAR receptors, a synthetic T cell receptor that coordinates a suite of immune responses, and SynNotch, a synthetic receptor that can be programmed to activate just about any gene. In a mouse model of cancer, treatment with SNAP-CAR T cells shrunk tumors and greatly prolonged survival, an important proof-of-concept that sets the stage to test this approach in clinical trials in partnership with Coeptis Therapeutics, which has licensed the SNAP-CAR technology from Pitt. The discovery could extend into solid tumors and give more patients access to the game-changing results CAR T cell therapy has produced in certain blood cancers. With the addition of SNAP, the possibilities for customized therapies become almost endless.
Adding a molecular anchor to the key protein used to recognize cancer in cellular immunotherapies can make the treatments significantly more effective.
BigField GEG Tech's insight:
CAR T cells have shown some success in the clinic in treating certain cancers, such as relapsed leukemia. However, CAR T cells have not been successful in delivering solid tumors, in part because of problems with immune cell activation. Adding a molecular anchor to the key protein used to recognize cancer in cellular immunotherapies can make treatments much more effective. The researchers found that immune cells with the anchored protein increased cancer killing, regardless of their cell type or the type of cancer targeted. The concept of molecular anchoring is thus a new design for improving chimeric antigen receptor (CAR) based immunotherapies. Anchored CARs have increased survival in animal models of several tumor types, including lung, bone and brain cancers. CARs have shown promise in the clinic, but have not yet achieved widespread success in all tumor types. The findings were published in Nature Biotechnology.
The efficacy of adoptive T cell therapies for cancer treatment can be limited by suppressive signals from both extrinsic factors and intrinsic inhibitory checkpoints1,2. Targeted gene editing has the potential to overcome these limitations and enhance T cell therapeutic function3–10. Here we performed multiple genome-wide CRISPR knock-out screens under different immunosuppressive conditions to identify genes that can be targeted to prevent T cell dysfunction. These screens converged on RASA2, a RAS GTPase-activating protein (RasGAP) that we identify as a signalling checkpoint in human T cells, which is downregulated upon acute T cell receptor stimulation and can increase gradually with chronic antigen exposure. RASA2 ablation enhanced MAPK signalling and chimeric antigen receptor (CAR) T cell cytolytic activity in response to target antigen. Repeated tumour antigen stimulations in vitro revealed that RASA2-deficient T cells show increased activation, cytokine production and metabolic activity compared with control cells, and show a marked advantage in persistent cancer cell killing. RASA2-knockout CAR T cells had a competitive fitness advantage over control cells in the bone marrow in a mouse model of leukaemia. Ablation of RASA2 in multiple preclinical models of T cell receptor and CAR T cell therapies prolonged survival in mice xenografted with either liquid or solid tumours. Together, our findings highlight RASA2 as a promising target to enhance both persistence and effector function in T cell therapies for cancer treatment. Genome-wide CRISPR screens, biochemical studies and animal models show that RASA2 has a key role in regulating T cell function and has potential as a genetic target for enhancing anti-tumour immunity.
BigField GEG Tech's insight:
T cells used in immunotherapy treatments can become exhausted by the task of fighting cancer cells or shut down when they enter tumours. However, a set of CRISPR screens allowed researchers to deactivate each gene in the genome, one at a time, in a pool of human T cells and the team found a handful of candidates that could make T cells resistant to key aspects of the immunosuppressive microenvironment often present in tumours. The researchers were particularly intrigued by a gene called RASA2, as it had never been associated with immune cell function before. The team created T cells with the RASA2 gene knocked out. They then subjected these T cells to various "stress tests" by repeatedly exposing them to cancer cells and models of the tumour microenvironment. They compared the performance of these cells to the original therapeutic T cells that still contained a functional RASA2 gene. Long after the original cells had lost their cancer-fighting abilities, the cells with knocked-out RASA2 remained remarkably tireless. The researchers thus made the therapeutic cells more resistant. This discovery could help overcome a major factor limiting the success of these promising therapies in the fight against solid and liquid tumours.

Synthetic receptor signalling has the potential to endow adoptively transferred T cells with new functions that overcome major barriers in the treatment of solid tumours, including the need for conditioning chemotherapy1,2. Here we designed chimeric receptors that have an orthogonal IL-2 receptor extracellular domain (ECD) fused with the intracellular domain (ICD) of receptors for common γ-chain (γc) cytokines IL-4, IL-7, IL-9 and IL-21 such that the orthogonal IL-2 cytokine elicits the corresponding γc cytokine signal. Of these, T cells that signal through the chimeric orthogonal IL-2Rβ-ECD–IL-9R-ICD (o9R) are distinguished by the concomitant activation of STAT1, STAT3 and STAT5 and assume characteristics of stem cell memory and effector T cells. Compared to o2R T cells, o9R T cells have superior anti-tumour efficacy in two recalcitrant syngeneic mouse solid tumour models of melanoma and pancreatic cancer and are effective even in the absence of conditioning lymphodepletion. Therefore, by repurposing IL-9R signalling using a chimeric orthogonal cytokine receptor, T cells gain new functions, and this results in improved anti-tumour activity for hard-to-treat solid tumours. Synthetic chimeric orthogonal IL-2 receptors that incorporate the intracellular domain of receptors for other γ-chain cytokines such as IL-9 can reroute orthogonal signalling and alter the phenotype of T cells to improve anti-tumour responses.
BigField GEG Tech's insight:
Researchers have shown that a synthetic IL-9 receptor allows anti-cancer T cells to do their job without the need for chemo or radiation. T cells modified with the synthetic IL-9 receptor were potent against tumours in mice, as published in Nature. This group of researchers were interested in testing modified versions of the synthetic receptor that transmit other cytokine signals from the common gamma chain family: IL-4, -7, -9 and -21. Of the synthetic common gamma chain signals, the IL-9 signal was worth studying and unlike other cytokines, IL-9 signalling is not generally active in naturally ocurring T cells. The synthetic IL-9 signal gave the T cells a unique blend of stem cell and killer cell qualities that made them more robust in fighting tumours. In particular, the researchers targeted two types of difficult-to-treat cancer models in mice: pancreatic cancer and melanoma. They used T cells targeted to the cancer cells via the natural T cell receptor or a chimeric antigen receptor (CAR). In all cases, T cells engineered with synthetic IL-9 receptor signalling were superior and helped cure some tumours in mice when they could not do otherwise.
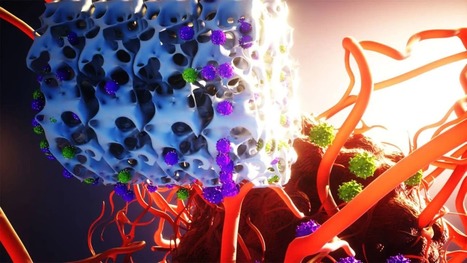
Despite their clinical success, chimeric antigen receptor (CAR)-T cell therapies for B cell malignancies are limited by lengthy, costly and labor-intensive ex vivo manufacturing procedures that might lead to cell products with heterogeneous composition. Here we describe an implantable Multifunctional Alginate Scaffold for T Cell Engineering and Release (MASTER) that streamlines in vivo CAR-T cell manufacturing and reduces processing time to a single day. When seeded with human peripheral blood mononuclear cells and CD19-encoding retroviral particles, MASTER provides the appropriate interface for viral vector-mediated gene transfer and, after subcutaneous implantation, mediates the release of functional CAR-T cells in mice. We further demonstrate that in vivo-generated CAR-T cells enter the bloodstream and control distal tumor growth in a mouse xenograft model of lymphoma, showing greater persistence than conventional CAR-T cells. MASTER promises to transform CAR-T cell therapy by fast-tracking manufacture and potentially reducing the complexity and resources needed for provision of this type of therapy. Implantable scaffolds rapidly generate and release anti-tumor CAR-T cells in mice.
BigField GEG Tech's insight:
Many people are excluded from CAR T cell-based treatments because of its cost. One reason for the high cost is that the manufacturing process is complex, time-consuming and must be individually tailored to each cancer patient. So to address this challenge, the researchers created a biotechnology called Multifunctional Alginate Scaffolds for T cell Engineering and Release (MASTER) that is a biocompatible sponge-like material. To begin treatment, the researchers isolate the patient's T cells and mix these naive T cells with the modified virus. The researchers pour this mixture onto MASTER, which absorbs it. MASTER is decorated with the antibodies that activate the T cells, so the cell activation process begins almost immediately. Meanwhile, MASTER is surgically implanted into the patient. After implantation, as the T cells become activated, they begin to respond to the modified viruses, which reprogram them into CAR-T cells. MASTER is also imbued with interleukin factors that promote cell proliferation. After implantation, these interleukins begin to leach out, promoting rapid proliferation of CAR-T cells. In a proof-of-concept study involving lymphoma in mice, researchers found that this treatment was faster and more effective than conventional CAR-T cell cancer treatment.
A collaborative study led by the Monash Biomedicine Discovery Institute has discovered a new immune checkpoint that may be exploited for cancer therapy.
BigField GEG Tech's insight:
A collaborative study led by the Monash Biomedicine Discovery Institute has discovered a new immune checkpoint that could be exploited for cancer treatment. The study shows that by inhibiting the protein tyrosine phosphatase PTP1B in T cells, the body's immune response to cancer can be mobilized, helping to suppress tumor growth. Indeed, this study showed that using a new drug candidate, the abundance of PTP1B in tumor-infiltrating T cells is increased, limiting the ability of T cells to attack tumor cells and fight cancer. These findings identified PTP1B as an intracellular brake, or checkpoint, reminiscent of the PD-1 cell surface checkpoint whose blockade has revolutionized cancer treatment. Furthermore, beyond the improved response to PD-1 blockade, the authors showed that inhibition of PTP1B also significantly improved the efficacy of cell-based therapies using CAR T cells. The authors demonstrate that deletion or inhibition of PTP1B can significantly improve the ability of CAR T cells to attack solid tumors in mice, including breast cancer.
B cell-activating factor (BAFF) binds the three receptors BAFF-R, BCMA, and TACI, predominantly expressed on mature B cells. Almost all B cell cancers are reported to express at least one of these receptors. Here we develop a BAFF ligand-based chimeric antigen receptor (CAR) and generate BAFF CAR-T cells using a non-viral gene delivery method. We show that BAFF CAR-T cells bind specifically to each of the three BAFF receptors and are effective at killing multiple B cell cancers, including mantle cell lymphoma (MCL), multiple myeloma (MM), and acute lymphoblastic leukemia (ALL), in vitro and in vivo using different xenograft models. Co-culture of BAFF CAR-T cells with these tumor cells results in induction of activation marker CD69, degranulation marker CD107a, and multiple proinflammatory cytokines. In summary, we report a ligand-based BAFF CAR-T capable of binding three different receptors, minimizing the potential for antigen escape in the treatment of B cell cancers. Antigen escape represents a potential drawback of chimeric antigen receptor T cell (CAR-T) therapy targeting a single tumor-associated antigen. To reduce the risk of antigen escape, here the authors report the design and characterization of a BAFF ligand CAR-T that can recognize three different receptors (BAFF-R, BCMA and TACI), demonstrating in vitro and in vivo cytotoxicity against multiple B cell cancer models.
BigField GEG Tech's insight:
Researchers at Seidman Cancer Center and Case Western Reserve University Hospitals have developed a new approach to CAR T cell therapy for B-cell cancers that triples targeted antigens on cancer cells. This approach promises to significantly reduce the potential for antigen escape currently found in CAR T therapies that target only CD19. The novel B-cell activating factor (BAFF) CAR T product developed here specifically binds to each of three receptors instead of one - BAFF-R, BCMA and TACI, providing more therapeutic options. At least two of these three receptors are found in almost all B-cell cancers, with some cancers expressing all three. Experimental results reported in Nature Communications show that BAFF CAR T is effective in killing several B-cell cancers. In addition, studies show robust in vitro and in vivo cytotoxicity exerted by CAR T BAFFs against mantle cell lymphoma, multiple myeloma, and mouse xenograft models of acute lymphoblastic leukemia. An Investigational New Drug application with the U.S. Food and Drug Administration will be filed in the coming weeks with Luminary Therapeutics and the team plans to initiate a clinical trial of BAFF CAR T therapy in patients with non-Hodgkin's lymphoma within the next few months.
Chimeric antigen receptor T-cell therapy followed by pembrolizumab appeared safe and feasible for patients with malignant pleural mesothelioma, phase 1 trial results published in Cancer Discovery showed.Eighty-three percent of patients who received the regimen remained alive 1 year after CAR-T infusion. Researchers reported no cases of high-grade cytokine release syndrome or neurotoxicity.
BigField GEG Tech's insight:
According to the results of the phase 1 trial published in Cancer Discovery, the combination of CAR T cell therapy and pembrolizumab, an anti-PD-1 therapy, appeared safe and feasible for patients with malignant pleural mesothelioma. 83% of patients who received the regimen remained alive 1 year after CAR-T infusion. The investigators reported no cases of high-grade cytokine release syndrome or neurotoxicity. This study was done with the use of a dose escalation of autologous CAR T cells that target the mesothelin protein on the surface of cancer cells and express an iCaspase-9 gene that serves as a "safety switch" to shut down the activity of the engineered T cells. The investigators chose a locoregional CAR-T dosing strategy because this type of cancer does not typically metastasize outside the chest cavity. A Phase 2 study using a fixed dose of mesothelin-directed CAR T cells followed by pembrolizumab is already underway. Four patients have been treated to date.

Researchers from Queen Mary University of London, have identified a protein that may represent a novel therapeutic target for the treatment of pancreatic cancer.
BigField GEG Tech's insight:
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer and has the lowest survival rate of all common cancers. Only about 7% of people diagnosed with this type of cancer in the UK survive their cancer for 5 years or more. However, a new protein called CEACAM7 has been identified by researchers at Queen Mary University. It could be a new therapeutic target for the treatment of PDAC which is the most common type of pancreatic cancer. In this study, the researchers developed a novel CAR T cell therapy using part of an anti-CEACAM7 antibody from Professor Brad Nelson (British Columbia, Canada). They then modified the killer T cells and presented on their surface this new CAR protein that recognizes and binds to CEACAM7, directing the killer T cells to kill only the cells with CEACAM7. Using this protein as a target, the researchers were able to create a CAR T cell therapy that killed pancreatic cancer cells in a pre-clinical model.
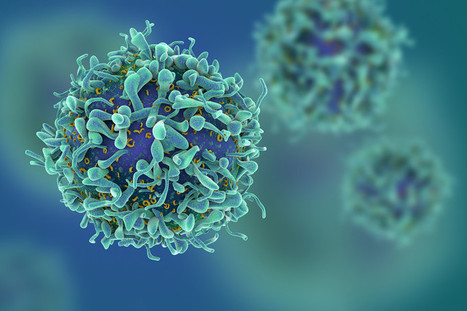
CAR T cells that target senescent cells combat disease in mouse models.
BigField GEG Tech's insight:
Senescence is a hallmark of cellular ageing and contributes to many diseases. A new method enabling immune cells to target senescent cells might offer improved therapeutic options.
|





Graft-versus-host disease occurs when a donor's cells attack the recipient's tissues, usually following a bone marrow or stem cell transplant. In a recent study, a new technique involving the combination of Chimeric Antigen Receptors (CARs) with Mesenchymal Stromal Cells (MSCs), resulting in modified stem cells known as CAR-MSCs, was used to specifically target a protein linked to graft-versus-host disease, but also to inflammatory bowel diseases such as ulcerative colitis and Crohn's disease. In mouse models, when stimulated by the specific protein for which they were designed, CAR-MSCs showed an enhanced ability to reach the inflamed area, better control inflammation and improve outcome and survival. This was mediated by a change in the genetic signature of CAR-MSCs, the proteins they released and receptor expression. These preliminary results pave the way for future applications of this technology, paving the way for improving the versatility of the therapy to treat various diseases across the autoimmune spectrum.