



Research and publish the best content.
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account
Already have an account: Login

Genetic Engineering Publications - GEG Tech top picks
36.6K views |
+0 today
 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
BigField GEG Tech's insight:
In this study the scientists show that the commonly used Cas9 can be modified to recognize alternative protospacer adjacent motif (PAM) sequences using structural information, bacterial selection-based directed evolution, and combinatorial design. These altered PAM specificity variants enable robust editing of endogenous gene sites in zebrafish and human cells and they identify and characterize a SpCas9 variant that exhibits improved specificity in human cells. 
|
BigField GEG Tech's insight:
Here, the scientists report that four to six bases at the 3′ end of the short CRISPR RNA (crRNA) used to program Cpf1 nucleases are insensitive to single base mismatches, but that many of the other bases in this region of the crRNA are highly sensitive to single or double substitutions. Using GUIDE-seq and targeted deep sequencing analyses performed with both Cpf1 nucleases, they were unable to detect off-target cleavage for more than half of 20 different crRNAs. Our results suggest that AsCpf1 and LbCpf1 are highly specific in human cells.
BigField GEG Tech's insight:
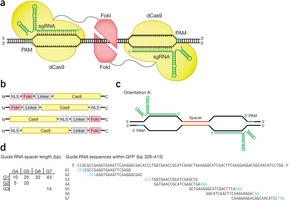
The fusion Cas9/FokI enhances specificity of CRISPR system without reduce the efficiency. In human cells, the authors shown that this fusion allows the modification of target DNA sites with a 140-fold higher specificity than wild-type Cas9 and with an efficiency similar to that of paired Cas9 'nickases'.
|




The scientists recently reported Digenome-seq (digested genome sequencing), a method for in vitro identification of potential off-target sites, and They evaluated the specificity of CRISPR–Cas9 and CRISPR–Cpf1 endonuclease by whole-genome sequencing. Digenome-seq pinpoints the exact location of double-strand break (DSB) sites by recognizing specific patterns of aligned reads. However, the analysis pipeline presented in their previous report required extensive manual interaction and produced several large intermediate files, resulting in a long running time. Here, the scientists present a redesigned analysis tool for Digenome-seq data that runs on web browsers. The core algorithm of the tool is written in C++ and compiled to asm.js (http://asmjs.org/), a preoptimized subset of JavaScript. Users can instantly perform the complete analysis in an ordinary web browser (Supplementary Note 1) with fast execution speed without uploading any data to a server and without local tool installation. In their benchmark, the full analysis for 100 GB of BAM file took 3 h for whole analysis on Intel i5 3570k central processing unit in a single thread.