Your new post is loading...
Your new post is loading...
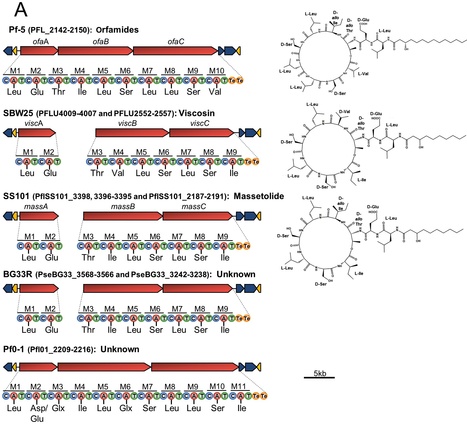
We provide here a comparative genome analysis of ten strains within the Pseudomonas fluorescens group including seven new genomic sequences. These strains exhibit a diverse spectrum of traits involved in biological control and other multitrophic interactions with plants, microbes, and insects. Multilocus sequence analysis placed the strains in three sub-clades, which was reinforced by high levels of synteny, size of core genomes, and relatedness of orthologous genes between strains within a sub-clade. The heterogeneity of the P. fluorescensgroup was reflected in the large size of its pan-genome, which makes up approximately 54% of the pan-genome of the genus as a whole, and a core genome representing only 45–52% of the genome of any individual strain. We discovered genes for traits that were not known previously in the strains, including genes for the biosynthesis of the siderophores achromobactin and pseudomonine and the antibiotic 2-hexyl-5-propyl-alkylresorcinol; novel bacteriocins; type II, III, and VI secretion systems; and insect toxins. Certain gene clusters, such as those for two type III secretion systems, are present only in specific sub-clades, suggesting vertical inheritance. Almost all of the genes associated with multitrophic interactions map to genomic regions present in only a subset of the strains or unique to a specific strain. To explore the evolutionary origin of these genes, we mapped their distributions relative to the locations of mobile genetic elements and repetitive extragenic palindromic (REP) elements in each genome. The mobile genetic elements and many strain-specific genes fall into regions devoid of REP elements (i.e., REP deserts) and regions displaying atypical tri-nucleotide composition, possibly indicating relatively recent acquisition of these loci. Collectively, the results of this study highlight the enormous heterogeneity of the P. fluorescens group and the importance of the variable genome in tailoring individual strains to their specific lifestyles and functional repertoire.
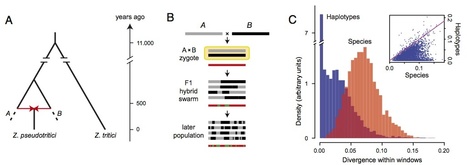
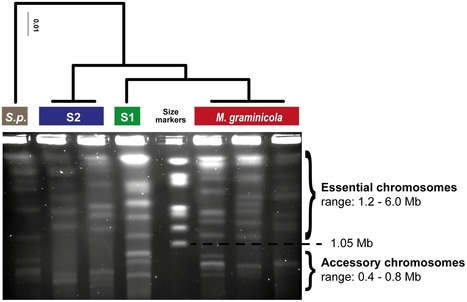
In a genome alignment of five individuals of the ascomycete fungus Zymoseptoria pseudotritici, a close relative of the wheat pathogen Z. tritici (synonym Mycosphaerella graminicola), we observed peculiar diversity patterns. Long regions up to 100 kb without variation alternate with similarly long regions of high variability. The variable segments in the genome alignment are organized into two main haplotype groups that have diverged ∼3% from each other. The genome patterns in Z. pseudotritici are consistent with a hybrid speciation event resulting from a cross between two divergent haploid individuals. The resulting hybrids formed the new species without backcrossing to the parents. We observe no variation in 54% of the genome in the five individuals and estimate a complete loss of variation for at least 30% of the genome in the entire species. A strong population bottleneck following the hybridization event caused this loss of variation. Variable segments in the Z. pseudotritici genome exhibit the two haplotypes contributed by the parental individuals. From our previously estimated recombination map of Z. tritici and the size distribution of variable chromosome blocks untouched by recombination we estimate that the hybridization occurred ∼380 sexual generations ago. We show that the amount of lost variation is explained by genetic drift during the bottleneck and by natural selection, as evidenced by the correlation of presence/absence of variation with gene density and recombination rate. The successful spread of this unique reproductively isolated pathogen highlights the strong potential of hybridization in the emergence of pathogen species with sexual reproduction.
The oomycete vegetable pathogen Phytophthora capsici has shown remarkable adaptation to fungicides and new hosts. Like other members of this destructive genus, P. capsici has an explosive epidemiology, rapidly producing massive numbers of asexual spores on infected hosts. In addition, P. capsici can remain dormant for years as sexually-recombined oospores, making it difficult to produce crops at infested sites, and allowing outcrossing populations to maintain significant genetic variation. Genome sequencing, development of a high-density genetic map, and integrative genomic/genetic characterization of P. capsici field isolates and intercross progeny revealed significant mitotic loss of heterozygosity (LOH) and higher levels of single nucleotide variants (SNVs) than those reported for humans, plants, and P. infestans. LOH was detected in clonally propagated field isolates and sexual progeny, cumulatively affecting >30% of the genome. LOH altered genotypes for more than 11,000 SNV sites and showed a strong association with changes in mating type and pathogenicity. Overall, it appears that LOH may provide a rapid mechanism for fixing alleles and may be an important component of adaptability for P. capsici.
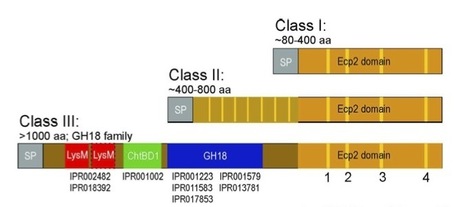
Most fungal plant pathogens secrete effector proteins during pathogenesis to manipulate their host’s defense and promote disease. These are so highly diverse in sequence and distribution, they are essentially considered as species-specific. However, we have recently shown the presence of homologous effectors in fungal species of the Dothideomycetes class. One such example is Ecp2, an effector originally described in the tomato pathogen Cladosporium fulvum but later detected in the plant pathogenic fungi Mycosphaerella fijiensis and Mycosphaerella graminicola as well. Here, using in silico sequence-similarity searches against a database of 135 fungal genomes and GenBank, we extend our queries for homologs of Ecp2 to the fungal kingdom and beyond, and further study their history of diversification. Our analyses show that Ecp2 homologs are members of an ancient and widely distributed superfamily of putative fungal effectors, which we term Hce2 for Homologs of C. fulvum Ecp2. Molecular evolutionary analyses show that the superfamily originated and diversified within the fungal kingdom, experiencing multiple lineage-specific expansions and losses that are consistent with the birth-and- death model of gene family evolution. Newly formed paralogs appear to be subject to diversification early after gene duplication events, whereas at later stages purifying selection acts to preserve diversity and the newly evolved putative functions. Some members of the Hce2 superfamily are fused to fungal Glycoside Hydrolase family 18 chitinases that show high similarity to the Zymocin killer toxin from the dairy yeast Kluyveromyces lactis, suggesting an analogous role in antagonistic interactions. The observed high rates of gene duplication and loss in the Hce2 superfamily, combined with diversification in both sequence and possibly functions within and between species suggest that Hce2s are involved in adaptation to stresses and new ecological niches. Such findings address the need to rationalize effector biology and evolution beyond the perspective of solely host-microbe interactions.
Meeting report of 2nd International Powdery Mildew Workshop and 3rd New Phytologist Workshop, in Zurich, Switzerland, February 2012.
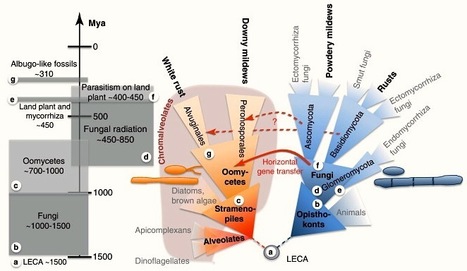
Although the oomycetes and fungi are evolutionarily very distantly related, both taxa evolved biotrophy on plant hosts several times independently, giving rise to rust- and mildew-like phenotypes. Differences in host colonization and adaptation may be reflected in genome size and by gain and loss of genes. In this opinion article we combine classical knowledge with recently sequenced pathogen genomes and present new hypotheses about the convergent evolution that led to these two distinct phenotypes in obligate biotrophs.
Worldwide, Dickeya (formerly Erwinia chrysanthemi) is causing soft rot diseases on a large diversity of crops and ornamental plants. Strains affecting potato are mainly found in D. dadantii, D. dianthicola and D. zeae, which appear to have a marked geographical distribution. Furthermore, a few Dickeya isolates from potato are attributed to D. chrysanthemi and D. dieffenbachiae. In Europe, isolates of Erwinia chrysanthemi biovar 1 and biovar 7 from potato are now classified in D. dianthicola. However, in the past few years, a new Dickeya biovar 3 variant, tentatively named ‘Dickeya solani’, has emerged as a common major threat, in particular in seed potatoes. Sequences of a fliC gene fragment were used to generate a phylogeny of Dickeya reference strains from culture collections and with this reference backbone, to classify pectinolytic isolates, i.e. Dickeya spp. from potato and ornamental plants. The reference strains of the currently recognized Dickeya species and ‘D. solani’ were unambiguously delineated in the fliC phylogram. D. dadantii, D. dianthicola and ‘D. solani’ displayed unbranched clades, while D. chrysanthemi, D. zeae and D. dieffenbachiae branched into subclades and lineages. Moreover, Dickeya isolates from diagnostic samples, in particular biovar 3 isolates from greenhouse ornamentals, formed several new lineages. Most of these isolates were positioned between the clade of ‘D. solani’ and D. dadantii as transition variants. New lineages also appeared in D. dieffenbachiae and in D. zeae. The strains and isolates of D. dianthicola and ‘D. solani’ were differentiated by a fliC sequence useful for barcode identification. A fliC TaqMan®real-time PCR was developed for ‘D. solani’ and the assay was provisionally evaluated in direct analysis of diagnostic potato samples. This molecular tool can support the efforts to control this particular phytopathogen in seed potato certification.
Microbiology Today: Genomics of emerging plant pathogens: too little, too late (2012)
Full article at http://www.sgm.ac.uk/pubs/micro_today/pdf/051214.pdf Last year during a visit to Colombia’s Zona Cafetera, my host singled out one coffee farm amid the enchanting rolling hills. That farm’s owner may have looked like the iconic Juan Valdez, but he is far from being cherished by his ‘cafeteros’ colleagues. He is infamous for having brought into Colombia a few coffee plants from Brazil. Unbeknown to him, a few leaves bore small orange spots, the telltale sign of the terrible coffee rust fungus, Hemileia vastatrix. Ever since that fateful introduction in 1983, Colombian cafeteros have struggled with managing this formidable foe. In recent years, after a brief lull, coffee rust came back with a vengeance casting a shadow on a critical Colombian agroindustry just as the country was emerging from years of social instability.
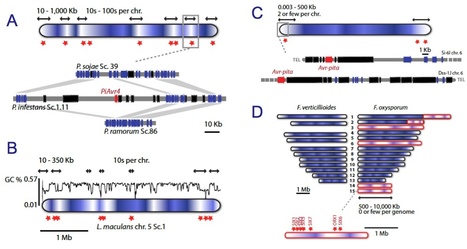
Many species of fungi and oomycetes are plant pathogens of great economic importance. Over the past 7 years, the genomes of more than 30 of these filamentous plant pathogens have been sequenced, revealing remarkable diversity in genome size and architecture. Whereas the genomes of many parasites and bacterial symbionts have been reduced over time, the genomes of several lineages of filamentous plant pathogens have been shaped by repeat-driven expansions. In these lineages, the genes encoding proteins involved in host interactions are frequently polymorphic and reside within repeat-rich regions of the genome. Here, we review the properties of these adaptable genome regions and the mechanisms underlying their plasticity, and we illustrate cases in which genome plasticity has contributed to the emergence of new virulence traits. We also discuss how genome expansions may have had an impact on the co-evolutionary conflict between these filamentous plant pathogens and their hosts.
Arbuscular mycorrhizal fungi (Glomeromycota) are the dominant symbionts of land plants and one of the oldest multicellular lineages that exist without evidence of sexual reproduction. The mechanisms that protect these organisms from extinction due to accumulation of deleterious mutations in the absence of sexual recombination are unclear. Glomeromycota reproduce by spores containing hundreds of nuclei, which represents a departure from the typical eukaryotic developmental pattern, where a multicellular organism is re-created from a uninucleate propagule. To understand whether the multinucleate spore makeup may have contributed to the evolutionary success of Glomeromycota, we examined the dynamics of spore nuclei in Glomus etunicatum using live three-dimensional imaging and mathematical models. We show that the spores are populated by an influx of a stream of nuclei from the surrounding mycelium rather than by divisions of a single founder nucleus. We present evidence that mechanisms of selection are likely to operate at the level of individual nuclei. On the basis of mathematical analyses of the effects that these nuclear dynamics have on the population mutation load, we postulate that the developmental patterns of sporogenesis have adaptive significance for moderating the accumulation of deleterious mutations and may have contributed to the evolutionary longevity of Glomeromycota.
Pathogens offer many of the most fascinating and well-studied examples of evolution because their speed of adaptation allows observation of evolutionary change within human lifetimes. The rapid evolution observed in pathogenic viruses, bacteria, and fungi affect human, animal, and plant health globally. For example, influenza viruses regularly recombine genes affecting host range, infection pathways, and virulence prior to the emergence of deadly outbreaks [1]. The emergence of antibiotic resistance in bacteria, including highly resistant Staphylococcus aureus strains and the re-emergence of resistant tuberculosis, is well documented [2], [3]. The recent outbreak in Africa of wheat stem rust caused by the Ug99 pathotype of the fungus Puccinia graminis tritici surprised many plant pathologists, who thought this ancient pathogen was defeated during the green revolution [4]. It is clear that pathogens engage host species in a constant race to evolve new defense mechanisms. By extension, we are constantly challenged to find new treatments and better containment measures to protect human health and essential crops.
The fungus Fusarium graminearum forms an intimate association with the host species wheat whilst infecting the floral tissues at anthesis. During the prolonged latent period of infection, extracellular communication between live pathogen and host cells must occur, implying a role for secreted fungal proteins. The wheat cells in contact with fungal hyphae subsequently die and intracellular hyphal colonisation results in the development of visible disease symptoms. Since the original genome annotation analysis was done in 2007, which predicted the secretome using TargetP, the F. graminearum gene call has changed considerably through the combined efforts of the BROAD and MIPS institutes. As a result of the modifications to the genome and the recent findings that suggested a role for secreted proteins in virulence, the F. graminearum secretome was revisited. In the current study, a refined F. graminearum secretome was predicted by combining several bioinformatic approaches. This strategy increased the probability of identifying truly secreted proteins. A secretome of 574 proteins was predicted of which 99% was supported by transcriptional evidence. The function of the annotated and unannotated secreted proteins was explored. The potential role(s) of the annotated proteins including, putative enzymes, phytotoxins and antifungals are discussed. Characterisation of the unannotated proteins included the analysis of Pfam domains and features associated with known fungal effectors, for example, small size, cysteine-rich and containing internal amino acid repeats. A comprehensive comparative genomic analysis involving 57 fungal and oomycete genomes revealed that only a small number of the predicted F. graminearum secreted proteins can be considered to be either species or sequenced strain specific.
Background - The potato genome sequence derived from the Solanum tuberosum Group Phureja clone DM1-3 516 R44 provides unparalleled insight into the genome composition and organisation of this important crop. A key class of genes that comprises the vast majority of plant resistance (R) genes contains a nucleotide-binding and leucine-rich repeat domain, and is collectively known as NB-LRRs. Results - As part of an effort to accelerate the process of functional R gene isolation, we performed an amino acid motif based search of the annotated potato genome and identified 438 NB-LRR type genes among the ~39,000 potato gene models. Of the predicted genes, 77 contain an N-terminal toll/interleukin 1 receptor (TIR)-like domain, and 107 of the remaining 361 non-TIR genes contain an N-terminal coiled-coil (CC) domain. Physical map positions were established for 370 predicted NB-LRR genes across all 12 potato chromosomes. The majority of NB-LRRs are physically organised within 63 identified clusters, of which 50 are homogeneous in that they contain NB-LRRs derived from a recent common ancestor. Conclusions - By establishing the phylogenetic and positional relationship of potato NB-LRRs, our analysis offers significant insight into the evolution of potato R genes. Furthermore, the data provide a blueprint for future efforts to identify and more rapidly clone functional NB-LRR genes from Solanum species.
|
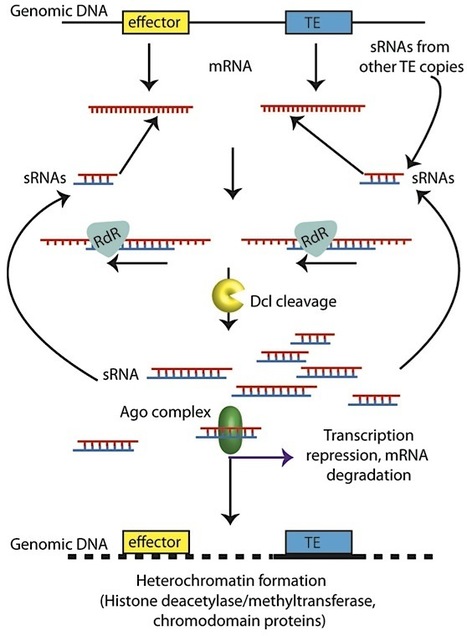
Transposable elements are ubiquitous residents in eukaryotic genomes. Often considered to be genomic parasites, they can lead to dramatic changes in genome organization, gene expression, and gene evolution. The oomycete plant pathogen Phytophthora infestans has evolved a genome organization where core biology genes are predominantly located in genome regions that have relatively few resident transposons. In contrast, disease effector-encoding genes are most frequently located in rapidly evolving genomic regions that are rich in transposons. P. infestans, as a eukaryote, likely uses RNA silencing to minimize the activity of transposons. We have shown that fusion of a short interspersed element (SINE) to an effector gene in P. infestans leads to the silencing of both the introduced fusion and endogenous homologous sequences. This is also likely to occur naturally in the genome of P. infestans, as transcriptional inactivation of effectors is known to occur, and over half of the translocated “RXLR class” of effectors are located within 2 kb of transposon sequences in the P. infestans genome. In this commentary, we review the diverse transposon inventory of P. infestans, its control by RNA silencing, and consequences for expression modulation of nearby effector genes in this economically important plant pathogen.
Cassava bacterial blight (CBB), incited by Xanthomonas axonopodis pv. manihotis (Xam), is the most important bacterial disease of cassava, a staple food source for millions of people in developing countries. Here we present a widely applicable strategy for elucidating the virulence components of a pathogen population. We report Illumina-based draft genomes for 65 Xam strains and deduce the phylogenetic relatedness of Xam across the areas where cassava is grown. Using an extensive database of effector proteins from animal and plant pathogens, we identify the effector repertoire for each sequenced strain and use a comparative sequence analysis to deduce the least polymorphic of the conserved effectors. These highly conserved effectors have been maintained over 11 countries, three continents, and 70 y of evolution and as such represent ideal targets for developing resistance strategies.
• Parasitism and saprotrophic wood decay are two fungal strategies fundamental for succession and nutrient cycling in forest ecosystems. An opportunity to assess the trade-off between these strategies is provided by the forest pathogen and wood decayer Heterobasidion annosum sensu lato.
• We report the annotated genome sequence and transcript profiling, as well as the quantitative trait loci mapping, of one member of the species complex: H. irregulare. Quantitative trait loci critical for pathogenicity, and rich in transposable elements, orphan and secreted genes, were identified.
• A wide range of cellulose-degrading enzymes are expressed during wood decay. By contrast, pathogenic interaction between H. irregulare and pine engages fewer carbohydrate-active enzymes, but involves an increase in pectinolytic enzymes, transcription modules for oxidative stress and secondary metabolite production.
• Our results show a trade-off in terms of constrained carbohydrate decomposition and membrane transport capacity during interaction with living hosts. Our findings establish that saprotrophic wood decay and necrotrophic parasitism involve two distinct, yet overlapping, processes.
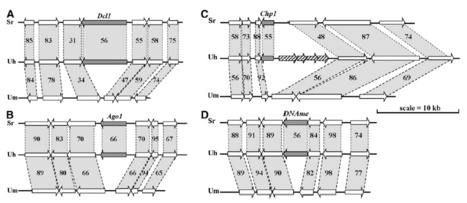
Ustilago hordei is a biotrophic parasite of barley (Hordeum vulgare). After seedling infection, the fungus persists in the plant until head emergence when fungal spores develop and are released from sori formed at kernel positions. The 26.1-Mb U. hordei genome contains 7113 protein encoding genes with high synteny to the smaller genomes of the related, maize-infecting smut fungi Ustilago maydis and Sporisorium reilianum but has a larger repeat content that affected genome evolution at important loci, including mating-type and effector loci. The U. hordei genome encodes components involved in RNA interference and heterochromatin formation, normally involved in genome defense, that are lacking in the U. maydis genome due to clean excision events. These excision events were possibly a result of former presence of repetitive DNA and of an efficient homologous recombination system in U. maydis. We found evidence of repeat-induced point mutations in the genome of U. hordei, indicating that smut fungi use different strategies to counteract the deleterious effects of repetitive DNA. The complement of U. hordei effector genes is comparable to the other two smuts but reveals differences in family expansion and clustering. The availability of the genome sequence will facilitate the identification of genes responsible for virulence and evolution of smut fungi on their respective hosts.
Links and references for the currently available fungal genome sequences or proposed (and austensibly funded) fungal genomes. Curated by the Stajich laboratory, UC Riverside http://fungalgenomes.org/
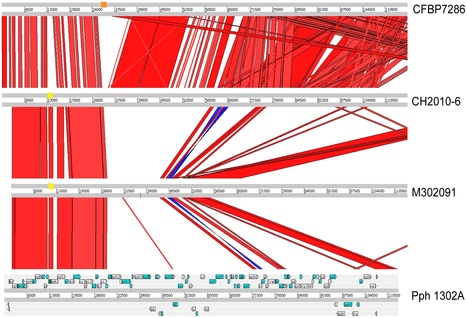
Intercontinental spread of emerging plant diseases is one of the most serious threats to world agriculture. One emerging disease is bacterial canker of kiwi fruit (Actinidia deliciosa and A. chinensis) caused by Pseudomonas syringae pv. actinidiae (PSA). The disease first occurred in China and Japan in the 1980s and in Korea and Italy in the 1990s. A more severe form of the disease broke out in Italy in 2008 and in additional countries in 2010 and 2011 threatening the viability of the global kiwi fruit industry. To start investigating the source and routes of international transmission of PSA, genomes of strains from China (the country of origin of the genus Actinidia), Japan, Korea, Italy and Portugal have been sequenced. Strains from China, Italy, and Portugal have been found to belong to the same clonal lineage with only 6 single nucleotide polymorphisms (SNPs) in 3,453,192 bp and one genomic island distinguishing the Chinese strains from the European strains. Not more than two SNPs distinguish each of the Italian and Portuguese strains from each other. The Japanese and Korean strains belong to a separate genetic lineage as previously reported. Analysis of additional European isolates and of New Zealand isolates exploiting genome-derived markers showed that these strains belong to the same lineage as the Italian and Chinese strains. Interestingly, the analyzed New Zealand strains are identical to European strains at the tested SNP loci but test positive for the genomic island present in the sequenced Chinese strains and negative for the genomic island present in the European strains. Results are interpreted in regard to the possible direction of movement of the pathogen between countries and suggest a possible Chinese origin of the European and New Zealand outbreaks.
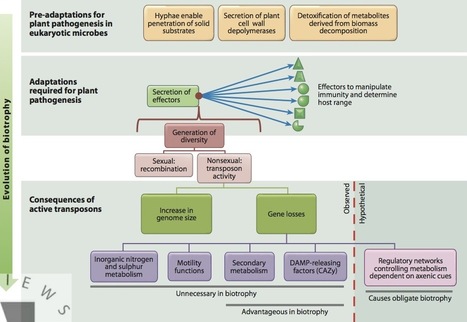
Biotrophy is a pervasive trait that evolved independently in plant pathogenic fungi and oomycetes. Comparative genomics of the first sequenced biotrophic pathogens highlight remarkable convergences, including gene losses in the metabolism of inorganic nitrogen, inorganic sulphur, and thiamine, and genes encoding carbohydrate active enzymes and secondary metabolism enzymes. Some biotrophs, but not all, display marked increases in overall genome size because of a proliferation of retrotransposons. I argue here that the release of constraints on transposon activity is driven by the advantages conferred by the genetic variability that results from transposition, in particular by the creation and diversification of broad palettes of effector genes. Increases in genome size and gene losses are the consequences of this trade-off. Genes that are not necessary for growth on a plant disappeared, but we still do not know what lost functions make some of these pathogens obligate.
A number of plant pathogenic fungi belonging to the genus Rhizopus are infamous for causing rice seedling blight. This plant disease is typically initiated by an abnormal swelling of the seedling roots without any sign of infection by the pathogen. This characteristic symptom is in fact caused by the macrocyclic polyketide metabolite rhizoxin that has been isolated from cultures of Rhizopus sp.. The phytotoxin exerts its destructive effect by binding to rice -tubulin, which results in inhibition of mitosis and cell cycle arrest. Owing to its remarkably strong antimitotic activity in most eukaryotic cells, including various human cancer cell lines, rhizoxin has attracted considerable interest as a potential antitumour drug. Here we show that rhizoxin is not biosynthesized by the fungus itself, but by endosymbiotic, that is, intracellular living, bacteria of the genus Burkholderia. Our unexpected findings unveil a remarkably complex symbiotic-pathogenic relationship that extends the fungus–plant interaction to a third, bacterial, key-player, and opens new perspectives for pest control.
Phytoplasmas are bacterial pathogens of plants that are transmitted by insects. These bacteria uniquely multiply intracellularly in both plants (Plantae) and insects (Animalia). Similarly to bacterial endosymbionts, phytoplasmas have reduced genomes with limited metabolic capabilities. Nonetheless, the chromosomes of many phytoplasmas are rich in repeated DNA consisting of mobile elements. Phytoplasmas produce an arsenal of effectors most of which are encoded on these mobile elements and on plasmids. These effectors target conserved plant transcription factors resulting in witches’ broom and leafy flower symptoms and suppression of plant defense to insect vectors that transmit the phytoplasmas. Future studies of these fascinating microbes will generate a wealth of new knowledge about forces that shape genomes and microbial interactions with multicellular hosts.
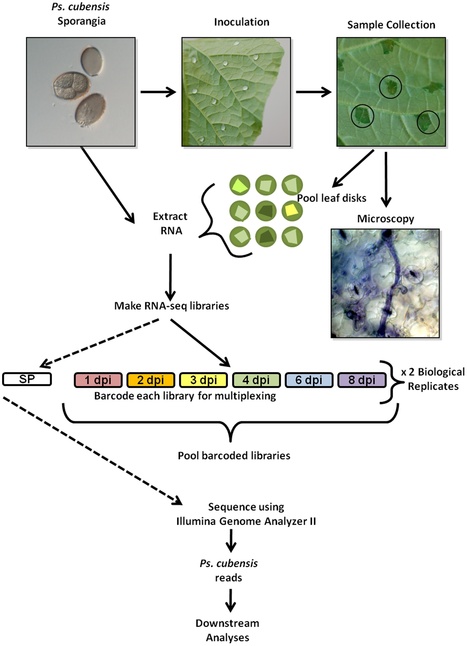
Pseudoperonospora cubensis, an oomycete, is the causal agent of cucurbit downy mildew, and is responsible for significant losses on cucurbit crops worldwide. While other oomycete plant pathogens have been extensively studied at the molecular level, Ps. cubensis and the molecular basis of its interaction with cucurbit hosts has not been well examined. Here, we present the first large-scale global gene expression analysis of Ps. cubensis infection of a susceptible Cucumis sativus cultivar, ‘Vlaspik’, and identification of genes with putative roles in infection, growth, and pathogenicity. Using high throughput whole transcriptome sequencing, we captured differential expression of 2383 Ps. cubensis genes in sporangia and at 1, 2, 3, 4, 6, and 8 days post-inoculation (dpi). Additionally, comparison of Ps. cubensis expression profiles with expression profiles from an infection time course of the oomycete pathogen Phytophthora infestans on Solanum tuberosum revealed similarities in expression patterns of 1,576–6,806 orthologous genes suggesting a substantial degree of overlap in molecular events in virulence between the biotrophic Ps. cubensis and the hemi-biotrophic P. infestans. Co-expression analyses identified distinct modules of Ps. cubensis genes that were representative of early, intermediate, and late infection stages. Collectively, these expression data have advanced our understanding of key molecular and genetic events in the virulence of Ps. cubensis and thus, provides a foundation for identifying mechanism(s) by which to engineer or effect resistance in the host.
Phytopathogens secrete effector proteins to manipulate their hosts for effective colonization. Hemibiotrophic fungi must maintain host viability during initial biotrophic growth and elicit host death for subsequent necrotrophic growth. To identify effectors mediating these opposing processes, we deeply sequenced the transcriptome of Colletotrichum higginsianum infecting Arabidopsis. Most effector genes are host-induced and expressed in consecutive waves associated with pathogenic transitions, indicating distinct effector suites are deployed at each stage. Using fluorescent protein tagging and transmission electron microscopy-immunogold labelling, we found effectors localised to stage-specific compartments at the host-pathogen interface. In particular, we show effectors are focally secreted from appressorial penetration pores before host invasion, revealing new levels of functional complexity for this fungal organ. Furthermore, we demonstrate that antagonistic effectors either induce or suppress plant cell death. Based on these results we conclude that hemibiotrophy in Colletotrichum is orchestrated through the coordinated expression of antagonistic effectors supporting either cell viability or cell death.
|