Your new post is loading...
Your new post is loading...
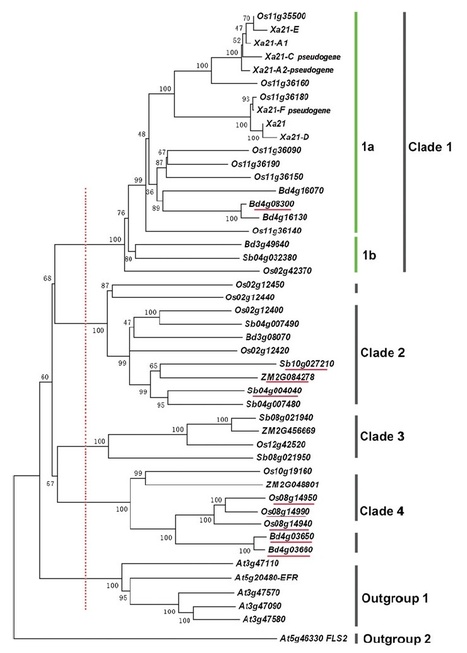
The XA21 protein has broad spectrum resistance against Xanthomonas oryzae pv. oryzae. Although Xa21-mediated immunity is well characterized, little is known about the origin and evolutionary history of this gene in grasses. Therefore, we analyzed all Xa21 gene homologs in eight whole-genome sequenced rice lines, as well as in four gramineous genomes, rice, Brachypodium, sorghum and maize; using Arabidopsis Xa21 homologs as outgroups, 17, 7, 7 and 3 Xa21 homologs were detected in these four grasses, respectively. Synteny and phylogenetic analysis showed that frequent gene translocation, duplication and/or loss, have occurred at Xa21 homologous loci, suggesting that they have undergone or are undergoing rapid generation of copy number variations. Within the rice species, the high level of nucleotide diversity between Xa21-like orthologs showed a strong association with the presence/absence haplotypes, suggesting that the genetic structure of rice lines plays an important role in the variations between these Xa21-like orthologs. Strongly positive selection was detected in the core region of the leucine-rich repeat domains of the Xa21 subclade among the rice lines, indicating that the rapid gene diversification of Xa21 homologs may be a strategy for a given species to adapt to the changing spectrum of species-specific pathogens.
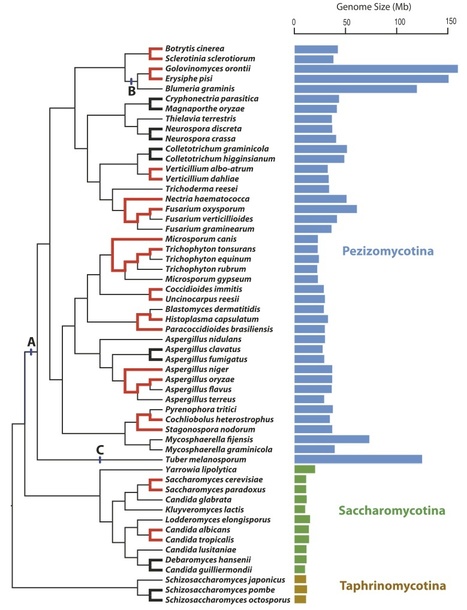
Fungi display a large diversity in genome size and complexity, variation that is often considered to be adaptive. But because non-adaptive processes can also have important consequences on the features of genomes, we investigated the relationship of genetic drift and genome size in the phylum Ascomycota using multiple indicators of genetic drift. We detected a complex relationship between genetic drift and genome size in fungi: genetic drift is associated with genome expansion on broad evolutionary time scales, as hypothesized for other eukaryotes; but within subphyla over smaller time scales, the opposite trend is observed. Moreover, fungi and bacteria display similar patterns of genome degradation that are associated with initial effects of genetic drift. We conclude that changes in genome size within Ascomycota have occurred using two different routes: large-scale genome expansions are catalyzed by increasing drift as predicted by the mutation-hazard model of genome evolution, and small-scale modifications in genome size are independent of drift.
Background - Eukaryotic genes with cyanobacterial ancestry in plastid-lacking protists have been regarded as important evolutionary markers implicating the presence of plastids in the early evolution of eukaryotes. Although recent genomic surveys demonstrated the presence of cyanobacterial and algal ancestry genes in the genomes of plastid-lacking protists, comparative analyses on the origin and distribution of those genes are still limited. Results - We identified 12 gene families with cyanobacterial ancestry in the genomes of a taxonomically wide range of plastid-lacking eukaryotes (Phytophthora [Chromalveolata], Naegleria [Excavata], Dictyostelium [Amoebozoa], Saccharomyces and Monosiga [Opisthokonta]) using a novel phylogenetic pipeline. The eukaryotic gene clades with cyanobacterial ancestry were mostly composed of genes from bikonts (Archaeplastida, Chromalveolata, Rhizaria and Excavata). We failed to find genes with cyanobacterial ancestry in Saccharomyces and Dictyostelium, except for a photorespiratory enzyme conserved among fungi. Meanwhile, we found several Monosiga genes with cyanobacterial ancestry, which were unrelated to other Opisthokonta genes. Conclusion - Our data demonstrate that a considerable number of genes with cyanobacterial ancestry have contributed to the genome composition of the plastid-lacking protists, especially bikonts. The origins of those genes might be due to lateral gene transfer events, or an ancient primary or secondary endosymbiosis before the diversification of bikonts. Our data also show that all genes identified in this study constitute multi-gene families with punctate distribution among eukaryotes, suggesting that the transferred genes could have survived through rounds of gene family expansion and differential reduction.
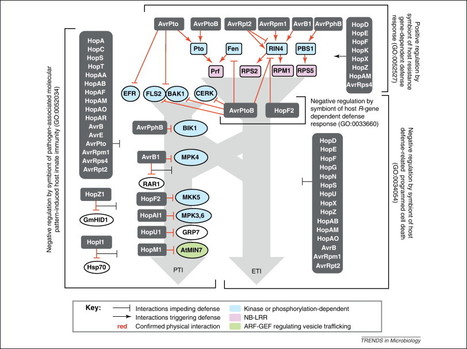
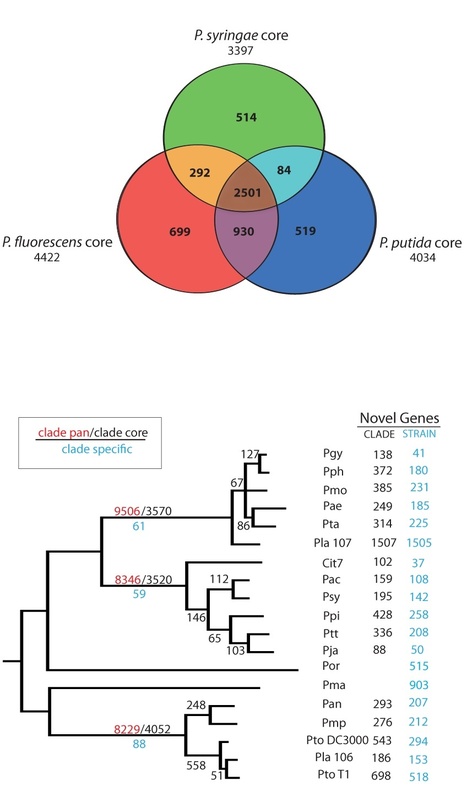
Many plant pathogens subvert host immunity by injecting compositionally diverse but functionally similar repertoires of cytoplasmic effector proteins. The bacterial pathogen Pseudomonas syringae is a model for exploring the functional structure of such repertoires. The pangenome of P. syringae encodes 57 families of effectors injected by the type III secretion system. Distribution of effector genes among phylogenetically diverse strains reveals a small set of core effectors targeting antimicrobial vesicle trafficking and a much larger set of variable effectors targeting kinase-based recognition processes. Complete disassembly of the 28-effector repertoire of a model strain and reassembly of a minimal functional repertoire reveals the importance of simultaneously attacking both processes. These observations, coupled with growing knowledge of effector targets in plants, support a model for coevolving molecular dialogs between effector repertoires and plant immune systems that emphasizes mutually-driven expansion of the components governing recognition.
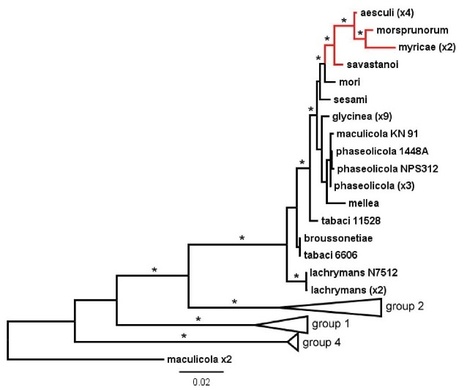
A recently emerging bleeding canker disease, caused by Pseudomonas syringae pathovar aesculi (Pae), is threatening European horse chestnut in northwest Europe. Very little is known about the origin and biology of this new disease. We used the nucleotide sequences of seven commonly used marker genes to investigate the phylogeny of three strains isolated recently from bleeding stem cankers on European horse chestnut in Britain (E-Pae). On the basis of these sequences alone, the E-Pae strains were identical to the Pae type-strain (I-Pae), isolated from leaf spots on Indian horse chestnut in India in 1969. The phylogenetic analyses also showed that Pae belongs to a distinct clade of P. syringae pathovars adapted to woody hosts. We generated genome-wide Illumina sequence data from the three E-Pae strains and one strain of I-Pae. Comparative genomic analyses revealed pathovar-specific genomic regions in Pae potentially implicated in virulence on a tree host, including genes for the catabolism of plant-derived aromatic compounds and enterobactin synthesis. Several gene clusters displayed intra-pathovar variation, including those encoding type IV secretion, a novel fatty acid biosynthesis pathway and a sucrose uptake pathway. Rates of single nucleotide polymorphisms in the four Pae genomes indicate that the three E-Pae strains diverged from each other much more recently than they diverged from I-Pae. The very low genetic diversity among the three geographically distinct E-Pae strains suggests that they originate from a single, recent introduction into Britain, thus highlighting the serious environmental risks posed by the spread of an exotic plant pathogenic bacterium to a new geographic location. The genomic regions in Pae that are absent from other P. syringae pathovars that infect herbaceous hosts may represent candidate genetic adaptations to infection of the woody parts of the tree.
Lettuce downy mildew (Bremia lactucae) is a rapidly adapting oomycete pathogen affecting commercial lettuce cultivation. Oomycetes are known to use a diverse arsenal of secreted proteins (effectors) to manipulate their hosts. Two classes of effector are known to be translocated by the host: the RXLRs and Crinklers. To gain insight into the repertoire of effectors used by B. lactucae to manipulate its host, we performed massively parallel sequencing of cDNA derived from B. lactucae spores and infected lettuce (Lactuca sativa) seedlings. From over 2.3 million 454 GS FLX reads, 59 618 contigs were assembled representing both plant and pathogen transcripts. Of these, 19 663 contigs were determined to be of B. lactucae origin as they matched pathogen genome sequences (SOLiD) that were obtained from >270 million reads of spore-derived genomic DNA. After correction of cDNA sequencing errors with SOLiD data, translation into protein models and filtering, 16 372 protein models remained, 1023 of which were predicted to be secreted. This secretome included elicitins, necrosis and ethylene-inducing peptide 1-like proteins, glucanase inhibitors and lectins, and was enriched in cysteine-rich proteins. Candidate host-translocated effectors included 78 protein models with RXLR effector features. In addition, we found indications for an unknown number of Crinkler-like sequences. Similarity clustering of secreted proteins revealed additional effector candidates. We provide a first look at the transcriptome of B. lactucae and its encoded effector arsenal.
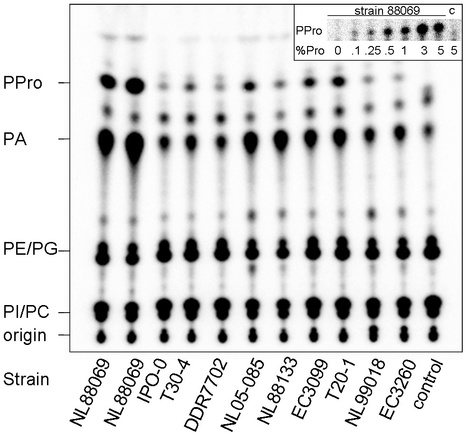
In eukaryotes phospholipase D (PLD) is involved in many cellular processes. Currently little is known about PLDs in oomycetes. Here we report that the oomycete plant pathogen Phytophthora infestans has a large repertoire of PLDs divided over six subfamilies: PXPH-PLD, PXTM-PLD, TM-PLD, PLD-likes, and type A and B sPLD-likes. Since the latter have signal peptides we developed a method using metabolically labelled phospholipids to monitor if P. infestans secretes PLD. In extracellular medium of ten P. infestans strains PLD activity was detected as demonstrated by the production of phosphatidic acid and the PLD specific marker phosphatidylalcohol.
The taxonomic class of oomycetes contains numerous pathogens of plants and animals, but is related to non-pathogenic diatoms and brown algae. Oomycetes have flexible genomes comprising large gene families that play roles in pathogenicity. The evolutionary processes that shaped the gene content have not yet been studied by applying systematic tree reconciliation of the phylome of these species. We analyzed evolutionary dynamics of ten Stramenopiles. Gene gains, duplications and losses were inferred by tree reconciliation of 18,459 gene trees constituting the phylome with a highly supported species phylogeny. We reconstructed a strikingly large last common ancestor of the Stramenopiles that contained ∼10,000 genes. Throughout evolution, the genomes of pathogenic oomycetes have constantly gained and lost genes, though gene gains through duplications outnumber the losses. The branch leading to the plant-pathogenic Phytophthora genus was identified as a major transition point characterized by increased frequency of duplication events that has likely driven the speciation within this genus. Large gene families encoding different classes of enzymes associated with pathogenicity such as glycoside hydrolases are formed by complex and distinct patterns of duplications and losses leading to their expansion in extant oomycetes. This study unveils the large-scale evolutionary dynamics that shaped the genomes of pathogenic oomycetes. By the application of phylogenetic based analyses methods it provides additional insights that shed light on the complex history of oomycete genome evolution and the emergence of large gene families characteristic for this important class of pathogens.
The obligate biotrophic rust fungus Melampsora larici-populina is the most devastating and widespread pathogen of poplars. Studies over recent years have identified various small secreted proteins (SSPs) from plant biotrophic filamentous pathogens and have highlighted their role as effectors in host-pathogen interactions. The recent analysis of the M. larici-populina genome sequence has revealed the presence of 1,184 SSP-encoding genes in this rust fungus. In the present study, the expression and evolutionary dynamics of these SSPs were investigated to pinpoint the arsenal of putative effectors that could be involved in the interaction between the rust fungus and poplar. Similarity with effectors previously described in Pucciniales, richness in cysteines and organization in large families were extensively detailed and discussed. Positive selection analyses conducted over clusters of paralogous genes revealed fast evolving candidate effectors. Transcript profiling of selected M. larici-populina SSPs showed a timely coordinated expression during leaf infection and the accumulation of four candidate effectors in distinct rust infection structures was demonstrated by immunolocalization. This integrated and multifaceted approach helps prioritizing candidate effector genes for functional studies.
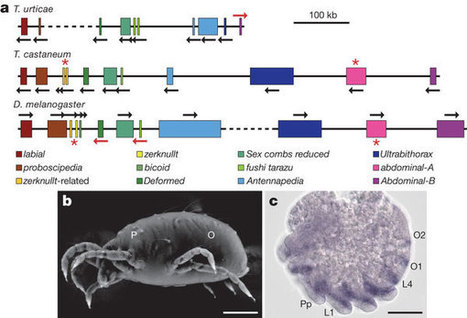
The spider mite Tetranychus urticae is a cosmopolitan agricultural pest with an extensive host plant range and an extreme record of pesticide resistance. Here we present the completely sequenced and annotated spider mite genome, representing the first complete chelicerate genome. At 90 megabases T. urticae has the smallest sequenced arthropod genome. Compared with other arthropods, the spider mite genome shows unique changes in the hormonal environment and organization of the Hox complex, and also reveals evolutionary innovation of silk production. We find strong signatures of polyphagy and detoxification in gene families associated with feeding on different hosts and in new gene families acquired by lateral gene transfer. Deep transcriptome analysis of mites feeding on different plants shows how this pest responds to a changing host environment. The T. urticae genome thus offers new insights into arthropod evolution and plant–herbivore interactions, and provides unique opportunities for developing novel plant protection strategies.
Understanding the molecular mechanisms of pathogen emergence is central to mitigating the impacts of novel infectious disease agents. The chytrid fungus Batrachochytrium dendrobatidis (Bd) is an emerging pathogen of amphibians that has been implicated in amphibian declines worldwide. Bd is the only member of its clade known to attack vertebrates. However, little is known about the molecular determinants of - or evolutionary transition to - pathogenicity in Bd. Here we sequence the genome of Bd's closest known relative - a non-pathogenic chytrid Homolaphlyctis polyrhiza (Hp). We first describe the genome of Hp, which is comparable to other chytrid genomes in size and number of predicted proteins. We then compare the genomes of Hp, Bd, and 19 additional fungal genomes to identify unique or recent evolutionary elements in the Bd genome. We identified 1,974 Bd-specific genes, a gene set that is enriched for protease, lipase, and microbial effector Gene Ontology terms. We describe significant lineage-specific expansions in three Bd protease families (metallo-, serine-type, and aspartyl proteases). We show that these protease gene family expansions occurred after the divergence of Bd and Hp from their common ancestor and thus are localized to the Bd branch. Finally, we demonstrate that the timing of the protease gene family expansions predates the emergence of Bd as a globally important amphibian pathogen.
Guest post by David Baltrus (@surt_lab) - I first want to thank Jonathan for giving me this opportunity. I am a big fan of “behind the science” stories, a habit I fed in grad school by reading every Perspectives (from the journal Genetics) article that I could get a hold of. Science can be rough, but I remember finding solace in stories about the false starts and triumphs of other researchers and how randomness and luck manage to figure into any discovery. If anything I hope to use this space to document this as it is fresh in my mind so that (inevitably) when the bad science days roll around I can have something to look back on. In the very least, I'm looking forward to mining this space in the future for quotes to prove just how little I truly understood about my research topics in 2011. It took a village to get this paper published, so apologies in advance to those that I fail to mention. Also want to mention this upfront, Marc Nishimura is my co-author and had a hand in every single aspect of this paper.
Recent sequencing projects have provided deep insight into fungal lifestyle-associated genomic adaptations. Here we report on the 25 Mb genome of the mutualistic root symbiont Piriformospora indica (Sebacinales, Basidiomycota) and provide a global characterization of fungal transcriptional responses associated with the colonization of living and dead barley roots. Extensive comparative analysis of the P. indica genome with other Basidiomycota and Ascomycota fungi that have diverse lifestyle strategies identified features typically associated with both, biotrophism and saprotrophism. The tightly controlled expression of the lifestyle-associated gene sets during the onset of the symbiosis, revealed by microarray analysis, argues for a biphasic root colonization strategy of P. indica. This is supported by a cytological study that shows an early biotrophic growth followed by a cell death-associated phase. About 10% of the fungal genes induced during the biotrophic colonization encoded putative small secreted proteins (SSP), including several lectin-like proteins and members of a P. indica-specific gene family (DELD) with a conserved novel seven-amino acids motif at the C-terminus. Similar to effectors found in other filamentous organisms, the occurrence of the DELDs correlated with the presence of transposable elements in gene-poor repeat-rich regions of the genome. This is the first in depth genomic study describing a mutualistic symbiont with a biphasic lifestyle. Our findings provide a significant advance in understanding development of biotrophic plant symbionts and suggest a series of incremental shifts along the continuum from saprotrophy towards biotrophy in the evolution of mycorrhizal association from decomposer fungi.
|
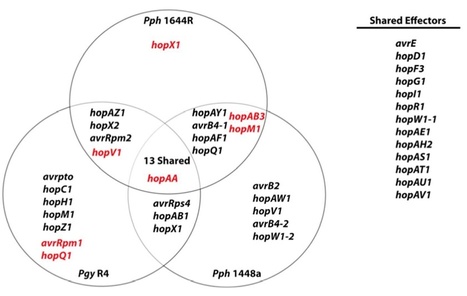
Biotrophic phytopathogens are typically limited to in their adapted host range. In recent decades, investigations have teased apart the general molecular basis of intraspecific variation for innate immunity of plants, typically involving receptor proteins that enable perception of Pathogen Associated Molecular Patterns (PAMPs) or avirulence elicitors from the pathogen as triggers for defense induction. However, teasing apart the molecular barriers that delimit host range between closely related phytopathogen isolates has been more difficult. Here, through genome comparisons and genetic manipulations, we investigate the underlying mechanisms that structure host range across closely related strains of Pseudomonas syringae isolated from different legume hosts. Although type III secretion-independent virulence genes are conserved across these three strains, we find that the presence of two genes encoding type III effectors (hopC1 and hopM1) and the absence of another (avrB2) potentially contribute to host range differences between pathovars glycinea and phaseolicola. These findings reinforce the idea that a complex genetic basis underlies host range evolution in plant pathogens. This complexity, likely the consequence of numerous evolutionary events, is present even in host-microbe interactions featuring relatively little divergence amongst both hosts and their adapted pathogens
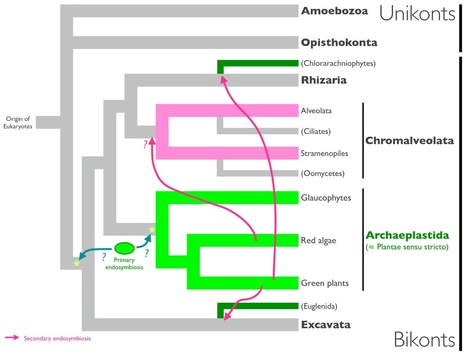
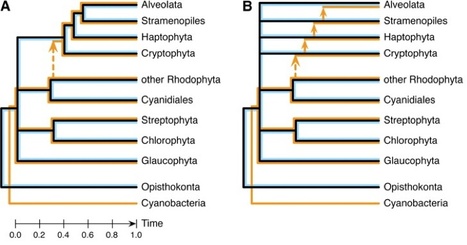
According to the chromalveolate hypothesis (Cavalier-Smith T. 1999. Principles of protein and lipid targeting in secondary symbiogenesis: euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J Eukaryot Microbiol 46:347–366), the four eukaryotic groups with chlorophyll c–containing plastids originate from a single photosynthetic ancestor, which acquired its plastids by secondary endosymbiosis with a red alga. So far, molecular phylogenies have failed to either support or disprove this view. Here, we devise a phylogenomic falsification of the chromalveolate hypothesis that estimates signal strength across the three genomic compartments: If the four chlorophyll c–containing lineages indeed derive from a single photosynthetic ancestor, then similar amounts of plastid, mitochondrial, and nuclear sequences should allow to recover their monophyly. Our results refute this prediction, with statistical support levels too different to be explained by evolutionary rate variation, phylogenetic artifacts, or endosymbiotic gene transfer. Therefore, we reject the chromalveolate hypothesis as falsified in favor of more complex evolutionary scenarios involving multiple higher order eukaryote–eukaryote endosymbioses.
Background - How photosynthetic organelles, or plastids, were acquired by diverse eukaryotes is among the most hotly debated topics in broad scale eukaryotic evolution. The history of plastid endosymbioses commonly is interpreted under the "chromalveolate" hypothesis, which requires numerous plastid losses from certain heterotrophic groups that now are entirely aplastidic. In this context, discoveries of putatively algal genes in plastid-lacking protists have been cited as evidence of gene transfer from a photosynthetic endosymbiont that subsequently was lost completely. Here we examine this evidence, as it pertains to the chromalveolate hypothesis, through genome-level statistical analyses of similarity scores from queries with two diatoms, Phaeodactylum tricornutum and Thalassiosira pseudonana, and two aplastidic sister taxa, Phytophthora ramorum and P. sojae. Results - Contingency tests of specific predictions of the chromalveolate model find no evidence for an unusual red algal contribution to Phytophthora genomes, nor that putative cyanobacterial sequences that are present entered these genomes through a red algal endosymbiosis. Examination of genes unrelated to plastid function provide extraordinarily significant support for both of these predictions in diatoms, the control group where a red endosymbiosis is known to have occurred, but none of that support is present in genes specifically conserved between diatoms and oomycetes. In addition, we uncovered a strong association between overall sequence similarities among taxa and relative sizes of genomic data sets in numbers of genes. Conclusion - Signal from "algal" genes in oomycete genomes is inconsistent with the chromalveolate hypothesis, and better explained by alternative models of sequence and genome evolution. Combined with the numerous sources of intragenomic phylogenetic conflict characterized previously, our results underscore the potential to be mislead by a posteriori interpretations of variable phylogenetic signals contained in complex genome-level data. They argue strongly for explicit testing of the different a priori assumptions inherent in competing evolutionary hypotheses.
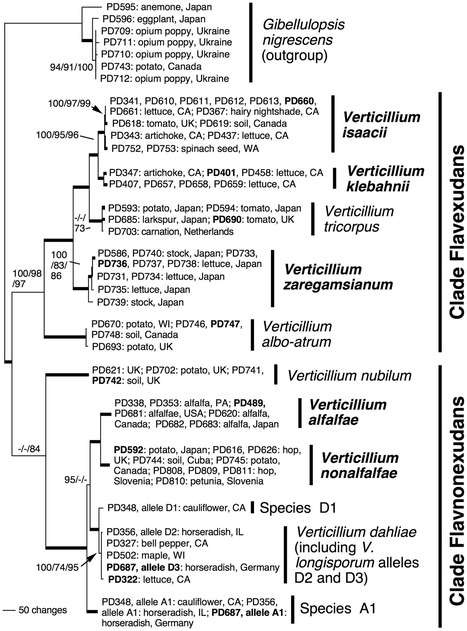
Knowledge of pathogen biology and genetic diversity is a cornerstone of effective disease management, and accurate identification of the pathogen is a foundation of pathogen biology. Species names provide an ideal framework for storage and retrieval of relevant information, a system that is contingent on a clear understanding of species boundaries and consistent species identification. Verticillium, a genus of ascomycete fungi, contains important plant pathogens whose species boundaries have been ill defined. Using phylogenetic analyses, morphological investigations and comparisons to herbarium material and the literature, we established a taxonomic framework for Verticillium comprising ten species, five of which are new to science. We used a collection of 74 isolates representing much of the diversity of Verticillium, and phylogenetic analyses based on the ribosomal internal transcribed spacer region (ITS), partial sequences of the protein coding genes actin (ACT), elongation factor 1-alpha (EF), glyceraldehyde-3-phosphate dehydrogenase (GPD) and tryptophan synthase (TS). Combined analyses of the ACT, EF, GPD and TS datasets recognized two major groups within Verticillium, Clade Flavexudans and Clade Flavnonexudans, reflecting the respective production and absence of yellow hyphal pigments. Clade Flavexudans comprised V. albo-atrum and V. tricorpus as well as the new species V. zaregamsianum, V. isaacii and V. klebahnii, of which the latter two were morphologically indistinguishable from V. tricorpus but may differ in pathogenicity. Clade Flavnonexudans comprised V. nubilum, V. dahliae and V. longisporum, as well as the two new species V. alfalfae and V. nonalfalfae, which resembled the distantly related V. albo-atrum in morphology. Apart from the diploid hybrid V. longisporum, each of the ten species corresponded to a single clade in the phylogenetic tree comprising just one ex-type strain, thereby establishing a direct link to a name tied to a herbarium specimen. A morphology-based key is provided for identification to species or species groups.
Pseudomonas savastanoi pv. savastanoi is a tumour-inducing pathogen of Olea europaea L. causing olive knot disease. Bioinformatic analysis of the draft genome sequence of strain NCPPB 3335, which encodes 5232 predicted coding genes on a total length of 5856 998 bp and a 57.12% G + C, revealed a large degree of conservation with Pseudomonas syringae pv. phaseolicola 1448A and P. syringae pv. tabaci 11528. However, NCPPB 3335 contains twelve variable genomic regions, which are absent in all previously sequenced P. syringae strains. Various features that could contribute to the ability of this strain to survive in a woody host were identified, including broad catabolic and transport capabilities for degrading plant-derived aromatic compounds, the duplication of sequences related to the biosynthesis of the phytohormone indoleacetic acid (iaaM, iaaH) and its amino acid conjugate indoleacetic acid-lysine (iaaL gene), and the repertoire of strain-specific putative type III secretion system effectors. Access to this seventh genome sequence belonging to the ‘P. syringae complex’ allowed us to identify 73 predicted coding genes that are NCPPB 3335-specific. Results shown here provide the basis for detailed functional analysis of a tumour-inducing pathogen of woody hosts and for the study of specific adaptations of a P. savastanoi pathovar.
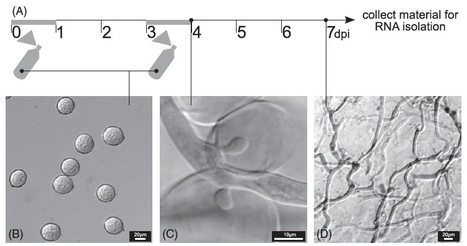
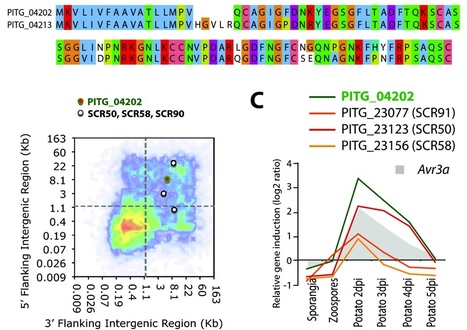
Background - Phytophthora infestans is the most devastating pathogen of potato and a model organism for the oomycetes. It exhibits high evolutionary potential and rapidly adapts to host plants. The P. infestans genome experienced a repeat-driven expansion relative to the genomes of Phytophthora sojae and Phytophthora ramorum and shows a discontinuous distribution of gene density. Effector genes, such as members of the RXLR and Crinkler (CRN) families, localize to expanded, repeat-rich and gene-sparse regions of the genome. This distinct genomic environment is thought to contribute to genome plasticity and host adaptation.
Results - We used in silico approaches to predict and describe the repertoire of P. infestans secreted proteins (the secretome). We defined the "plastic secretome" as a subset of the genome that (i) encodes predicted secreted proteins, (ii) is excluded from genome segments orthologous to the P. sojae and P. ramorum genomes and (iii) is encoded by genes residing in gene sparse regions of P. infestans genome. Although including only ~3% of P. infestans genes, the plastic secretome contains ~62% of known effector genes and shows >2 fold enrichment in genes induced in planta. We highlight 19 plastic secretome genes induced in planta but distinct from previously described effectors. This list includes a trypsin-like serine protease, secreted oxidoreductases, small cysteine-rich proteins and repeat containing proteins that we propose to be novel candidate virulence factors.
Conclusions - This work revealed a remarkably diverse plastic secretome. It illustrates the value of combining genome architecture with comparative genomics to identify novel candidate virulence factors from pathogen genomes.
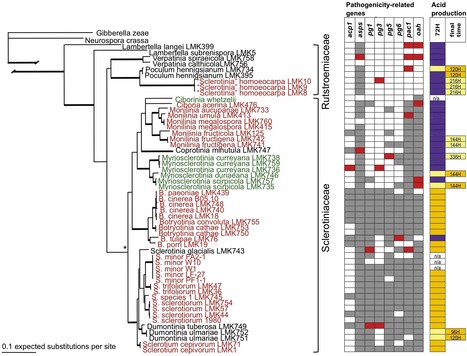
The Sclerotiniaceae (Ascomycotina, Leotiomycetes) is a relatively recently evolved lineage of necrotrophic host generalists, and necrotrophic or biotrophic host specialists, some latent or symptomless. We hypothesized that they inherited a basic toolbox of genes for plant symbiosis from their common ancestor. Maintenance and evolutionary diversification of symbiosis could require selection on toolbox genes or on timing and magnitude of gene expression. The genes studied were chosen because their products have been previously investigated as pathogenicity factors in the Sclerotiniaceae. They encode proteins associated with cell wall degradation: acid protease 1 (acp1), aspartyl protease (asps), and polygalacturonases (pg1, pg3, pg5, pg6), and the oxalic acid (OA) pathway: a zinc finger transcription factor (pac1), and oxaloacetate acetylhydrolase (oah), catalyst in OA production, essential for full symptom production in Sclerotinia sclerotiorum. Site-specific likelihood analyses provided evidence for purifying selection in all 8 pathogenicity-related genes. Consistent with an evolutionary arms race model, positive selection was detected in 5 of 8 genes. Only generalists produced large, proliferating disease lesions on excised Arabidopsis thaliana leaves and oxalic acid by 72 hours in vitro. In planta expression of oah was 10–300 times greater among the necrotrophic host generalists than necrotrophic and biotrophic host specialists; pac1 was not differentially expressed. Ability to amplify 6/8 pathogenicity related genes and produce oxalic acid in all genera are consistent with the common toolbox hypothesis for this gene sample. That our data did not distinguish biotrophs from necrotrophs is consistent with 1) a common toolbox based on necrotrophy and 2) the most conservative interpretation of the 3-locus housekeeping gene phylogeny – a baseline of necrotrophy from which forms of biotrophy emerged at least twice. Early oah overexpression likely expands the host range of necrotrophic generalists in the Sclerotiniaceae, while specialists and biotrophs deploy oah, or other as-yet-unknown toolbox genes, differently.
Rust fungi are obligate biotrophic pathogens that cause considerable damage on crop plants. Puccinia graminis f. sp. tritici, the causal agent of wheat stem rust, and Melampsora larici-populina, the poplar leaf rust pathogen, have strong deleterious impacts on wheat and poplar wood production, respectively. Filamentous pathogens such as rust fungi secrete molecules called disease effectors that act as modulators of host cell physiology and can suppress or trigger host immunity. Current knowledge on effectors from other filamentous plant pathogens can be exploited for the characterisation of effectors in the genome of recently sequenced rust fungi. We designed a comprehensive in silico analysis pipeline to identify the putative effector repertoire from the genome of two plant pathogenic rust fungi. The pipeline is based on the observation that known effector proteins from filamentous pathogens have at least one of the following properties: (i) contain a secretion signal, (ii) are encoded by in planta induced genes, (iii) have similarity to haustorial proteins, (iv) are small and cysteine rich, (v) contain a known effector motif or a nuclear localization signal, (vi) are encoded by genes with long intergenic regions, (vii) contain internal repeats, and (viii) do not contain PFAM domains, except those associated with pathogenicity. We used Markov clustering and hierarchical clustering to classify protein families of rust pathogens and rank them according to their likelihood of being effectors. Using this approach, we identified eight families of candidate effectors that we consider of high value for functional characterization. This study revealed a diverse set of candidate effectors, including families of haustorial expressed secreted proteins and small cysteine-rich proteins. This comprehensive classification of candidate effectors from these devastating rust pathogens is an initial step towards probing plant germplasm for novel resistance components.
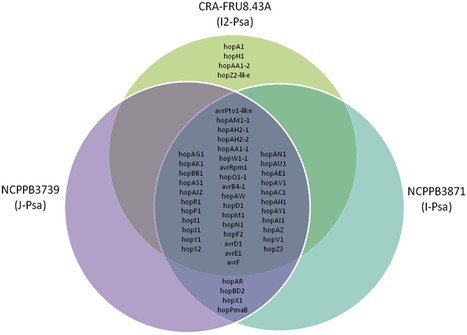
A recent re-emerging bacterial canker disease incited by Pseudomonas syringae pv. actinidiae (Psa) is causing severe economic losses to Actinidia chinensis and A. deliciosa cultivations in southern Europe, New Zealand, Chile and South Korea. Little is known about the genetic features of this pathovar. We generated genome-wide Illumina sequence data from two Psa strains causing outbreaks of bacterial canker on the A. deliciosa cv. Hayward in Japan (J-Psa, type-strain of the pathovar) and in Italy (I-Psa) in 1984 and 1992, respectively as well as from a Psa strain (I2-Psa) isolated at the beginning of the recent epidemic on A. chinensis cv. Hort16A in Italy. All strains were isolated from typical leaf spot symptoms. The phylogenetic relationships revealed that Psa is more closely related to P. s. pv. theae than to P. avellanae within genomospecies 8. Comparative genomic analyses revealed both relevant intrapathovar variations and putative pathovar-specific genomic regions in Psa. The genomic sequences of J-Psa and I-Psa were very similar. Conversely, the I2-Psa genome encodes four additional effector protein genes, lacks a 50 kb plasmid and the phaseolotoxin gene cluster, argK-tox but has acquired a 160 kb plasmid and putative prophage sequences. Several lines of evidence from the analysis of the genome sequences support the hypothesis that this strain did not evolve from the Psa population that caused the epidemics in 1984–1992 in Japan and Italy but rather is the product of a recent independent evolution of the pathovar actinidiae for infecting Actinidia spp. All Psa strains share the genetic potential for copper resistance, antibiotic detoxification, high affinity iron acquisition and detoxification of nitric oxide of plant origin. Similar to other sequenced phytopathogenic pseudomonads associated with woody plant species, the Psa strains isolated from leaves also display a set of genes involved in the catabolism of plant-derived aromatic compounds.
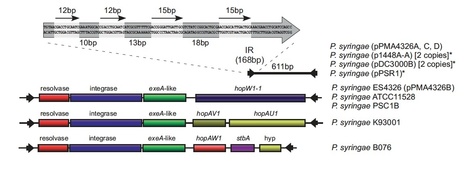
Miniature inverted terminal-repeat elements (MITEs) are non-autonomous mobile elements that have a significant impact on bacterial evolution. Here we characterize E622, a 611 bp virulence-associated MITE from Pseudomonas syringae, which contains no coding region, but has almost perfect 168 bp inverted repeats. Using an antibiotic-coupling assay, we show that E622 is transposable, and can mobilize an antibiotic resistance gene contained between its borders. Its predicted parent element, designated TnE622, has a typical transposon structure with a three-gene operon consisting of a resolvase, integrase, and exeA-like genes, which is bounded by the same terminal inverted repeats as E622. A broader genome-level survey of the E622/TnE622 inverted repeats identified homologs in Pseudomonas, Salmonella, Shewanella, Erwinia, Pantoea, and the cyanobacteria, Nostoc and Cyanothece, many of which appear to encompass known virulence genes, including toxins, enzymes, and type III secreted effectors. Its association with niche-specific genetic determinants, along with its persistence and evolutionary diversification indicates that this mobile element family has played a prominent role in the evolution of many agriculturally and clinically relevant pathogenic bacteria.
The fungus Mycosphaerella graminicola emerged as a new pathogen of cultivated wheat during its domestication ∼11,000 yr ago. We assembled 12 high-quality full genome sequences to investigate the genetic footprints of selection in this wheat pathogen and closely related sister species that infect wild grasses. We demonstrate a strong effect of natural selection in shaping the pathogen genomes with only ∼3% of nonsynonymous mutations being effectively neutral. Forty percent of all fixed nonsynonymous substitutions, on the other hand, are driven by positive selection. Adaptive evolution has affected M. graminicola to the highest extent, consistent with recent host specialization. Positive selection has prominently altered genes encoding secreted proteins and putative pathogen effectors supporting the premise that molecular host–pathogen interaction is a strong driver of pathogen evolution. Recent divergence between pathogen sister species is attested by the high degree of incomplete lineage sorting (ILS) in their genomes. We exploit ILS to generate a genetic map of the species without any crossing data, document recent times of species divergence relative to genome divergence, and show that gene-rich regions or regions with low recombination experience stronger effects of natural selection on neutral diversity. Emergence of a new agricultural host selected a highly specialized and fast-evolving pathogen with unique evolutionary patterns compared with its wild relatives. The strong impact of natural selection, we document, is at odds with the small effective population sizes estimated and suggest that population sizes were historically large but likely unstable.
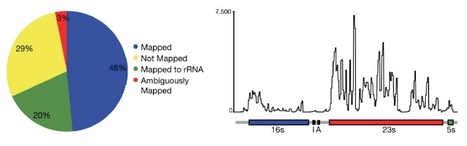
The throughput and single-base resolution of RNA-Sequencing (RNA-Seq) have contributed to a dramatic change in transcriptomic-based inquiries and resulted in many new insights into the complexities of bacterial transcriptomes. RNA-Seq could contribute to similar advances in our understanding of plant pathogenic bacteria but it is still a technology under development with limitations and unknowns that need to be considered. Here, we review some new developments for RNA-Seq and highlight recent findings for host-associated bacteria. We also discuss the technical and statistical challenges in the practical application of RNA-Seq for studying bacterial transcriptomes and describe some of the currently available solutions.
|